Publications
Peer-reviewed publications listed in reverse chronological order.
2025
- Mol Neurobiol
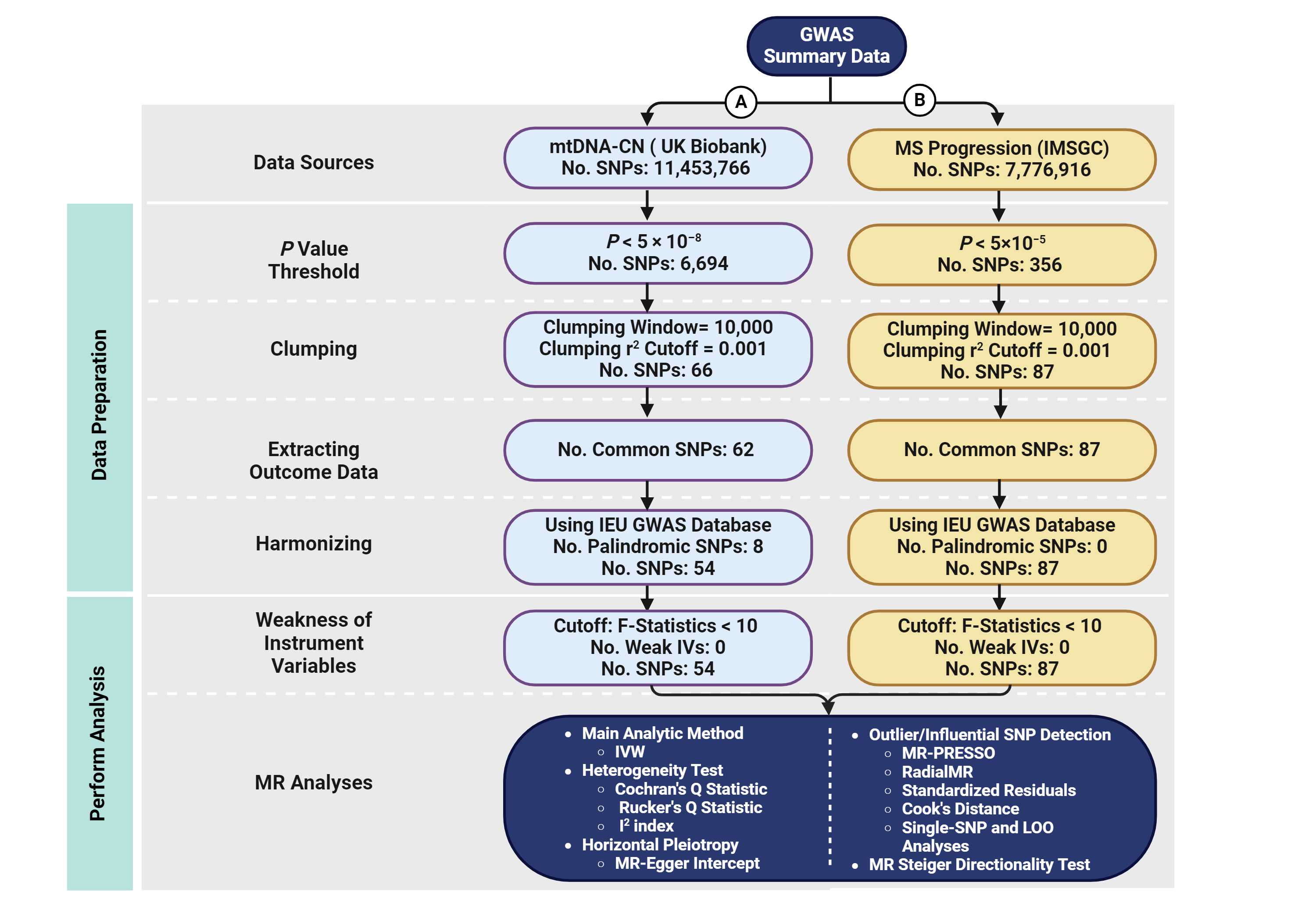 Mitochondrial DNA Copy Number as a Hidden Player in the Progression of Multiple Sclerosis: A Bidirectional Two-Sample Mendelian Randomization StudyHani Sabaie, Ali Taghavi Rad, Motahareh Shabestari, and 14 more authors2025
Mitochondrial DNA Copy Number as a Hidden Player in the Progression of Multiple Sclerosis: A Bidirectional Two-Sample Mendelian Randomization StudyHani Sabaie, Ali Taghavi Rad, Motahareh Shabestari, and 14 more authors2025The relationship between mitochondrial DNA copy number (mtDNA-CN) and multiple sclerosis (MS) progression remains unclear, as previous observational studies have reported conflicting results. This study aimed to clarify the association between mtDNA-CN and MS progression using a bidirectional two-sample Mendelian randomization (MR) approach. MR analyses were conducted using the latest summary statistics from genome-wide association studies (GWAS) on mtDNA-CN and MS progression. Single-nucleotide polymorphisms (SNPs) associated with mtDNA-CN were extracted from 383,476 participants of European ancestry in the UK Biobank, while SNPs associated with MS severity were obtained from the International Multiple Sclerosis Genetics Consortium (IMSGC), comprising 12,584 cases of European ancestry. The inverse variance weighted (IVW) method was used as the primary analysis. Potential heterogeneity and pleiotropy were evaluated, and sensitivity analyses were performed to ensure the robustness of the results. The forward MR analysis using the IVW method revealed no significant association between mtDNA-CN and MS progression (P = 0.487). However, reverse MR analysis identified a causal association between MS progression and mtDNA-CN (β = − 0.010, 95% CI = − 0.019 to − 0.001, P = 0.036). No evidence of heterogeneity or horizontal pleiotropy was found in the analyses. Sensitivity analyses yielded consistent results. Our findings suggest that MS progression may causally influence mtDNA-CN, highlighting the crucial role of mitochondria in the pathophysiology of MS. However, further research is needed to confirm mtDNA-CN as a reliable biomarker and a deeper understanding of the molecular mechanisms is necessary to develop targeted therapeutic interventions.
- Mult Scler Relat Disord
 Deciphering the bidirectional impact of leukocyte telomere length on multiple sclerosis progression: A Mendelian randomization studyHani Sabaie, Ali Taghavi Rad, Motahareh Shabestari, and 6 more authors2025
Deciphering the bidirectional impact of leukocyte telomere length on multiple sclerosis progression: A Mendelian randomization studyHani Sabaie, Ali Taghavi Rad, Motahareh Shabestari, and 6 more authors2025Observational studies have suggested a link between leukocyte telomere length (LTL) and multiple sclerosis (MS) progression, but the causal relationship remains uncertain. This study investigates the causal association between LTL and MS progression using a bidirectional two-sample Mendelian randomization (MR) approach. We analyzed genome-wide association summary statistics data from 472,174 individuals for LTL and 12,584 MS patients for disease progression. The primary method was the inverse variance weighted (IVW) approach, supported by sensitivity analyses to ensure robustness. The forward analysis revealed a significant positive causal relationship between LTL and MS progression (β = 0.107, 95 % CI = 0.006 to 0.209, P = 0.037). Conversely, the reverse analysis indicated a negative causal relationship (β = -0.010, 95 % CI = -0.020 to -0.001, P = 0.037). No heterogeneity or horizontal pleiotropy was found, and the sensitivity analyses confirmed consistent results. These findings suggest that telomere dynamics play a complex role in MS progression and highlight their potential as therapeutic targets. Further research is essential to uncover the biological mechanisms underlying the influence of telomeres on MS progression.
2024
- Mol Biol Rep
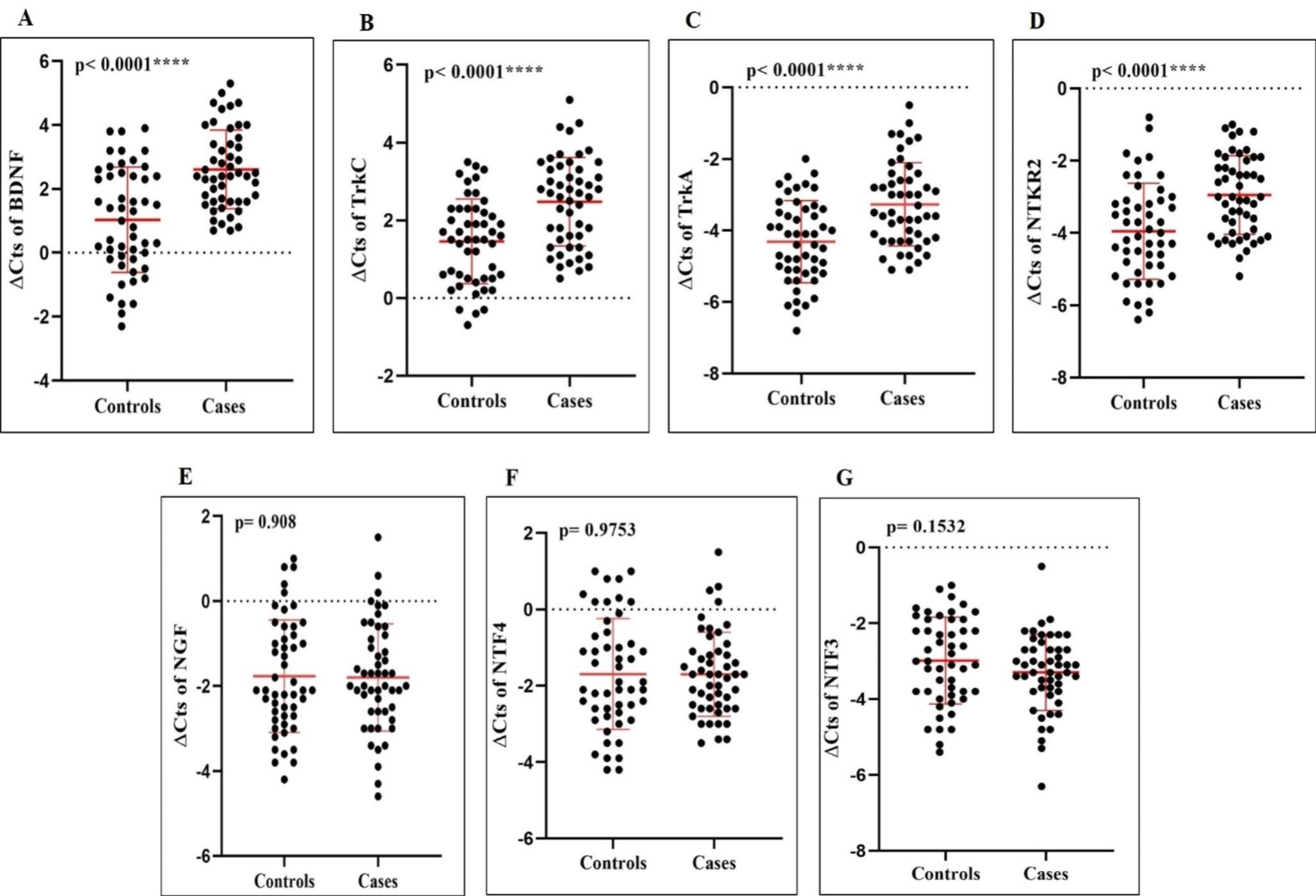 Neurotrophin growth factors and their receptors as promising blood biomarkers for Alzheimer’s Disease: a gene expression analysis studyMohammad Reza Asadi, Jalal Gharesouran, Hani Sabaie, and 7 more authors2024
Neurotrophin growth factors and their receptors as promising blood biomarkers for Alzheimer’s Disease: a gene expression analysis studyMohammad Reza Asadi, Jalal Gharesouran, Hani Sabaie, and 7 more authors2024Background Alzheimer’s disease (AD) is a multifaceted neurological ailment affecting more than 50 million individuals globally, distinguished by a deterioration in memory and cognitive abilities. Investigating neurotrophin growth factors could offer significant contributions to understanding AD progression and prospective therapeutic interventions. Methods and results The present investigation collected blood samples from 50 patients diagnosed with AD and 50 healthy individuals serving as controls. The mRNA expression levels of neurotrophin growth factors and their receptors were measured using quantitative PCR. A Bayesian regression model was used in the research to assess the relationship between gene expression levels and demographic characteristics such as age and gender. The correlations between variables were analyzed using Spearman correlation coefficients, and the diagnostic potential was assessed using a Receiver Operating Characteristic curve. NTRK2, TrkA, TrkC, and BDNF expression levels were found to be considerably lower (p-value < 0.05) in the blood samples of AD patients compared to the control group. The expression of BDNF exhibited the most substantial decrease in comparison to other neurotrophin growth factors. Correlation analysis indicates a statistically significant positive association between the genes. The ROC analysis showed that BDNF exhibited the greatest Area Under the Curve (AUC) value of 0.76, accompanied by a sensitivity of 70% and specificity of 66%. TrkC, TrkA, and NTRK2 demonstrated considerable diagnostic potential in distinguishing between cases and controls. Conclusion The observed decrease in the expression levels of NTRK2, TrkA, TrkC, and BDNF in AD patients, along with the identified associations between specific genes and their diagnostic capacity, indicate that these expressions have the potential to function as biomarkers for the diagnosis and treatment of AD.
- Gene
 A comprehensive review of the applications of RNA sequencing in celiac disease researchMaryam Shoaran, Hani Sabaie, Mehrnaz Mostafavi, and 1 more author2024
A comprehensive review of the applications of RNA sequencing in celiac disease researchMaryam Shoaran, Hani Sabaie, Mehrnaz Mostafavi, and 1 more author2024RNA sequencing (RNA-seq) has undergone substantial advancements in recent decades and has emerged as a vital technique for profiling the transcriptome. The transition from bulk sequencing to single-cell and spatial approaches has facilitated the achievement of higher precision at cell resolution. It provides valuable biological knowledge about individual immune cells and aids in the discovery of the molecular mechanisms that contribute to the development of autoimmune diseases. Celiac disease (CeD) is an autoimmune disorder characterized by a strong immune response to gluten consumption. RNA-seq has led to significantly advanced research in multiple fields, particularly in CeD research. It has been instrumental in studies involving comparative transcriptomics, nutritional genomics and wheat research, cancer research in the context of CeD, genetic and noncoding RNA-mediated epigenetic insights, disease monitoring and biomarker discovery, regulation of mitochondrial functions, therapeutic target identification and drug mechanism of action, dietary factors, immune cell profiling and the immune landscape. This review offers a comprehensive examination of recent RNA-seq technology research in the field of CeD, highlighting future challenges and opportunities for its application.
2023
- Front Cell Neurosci
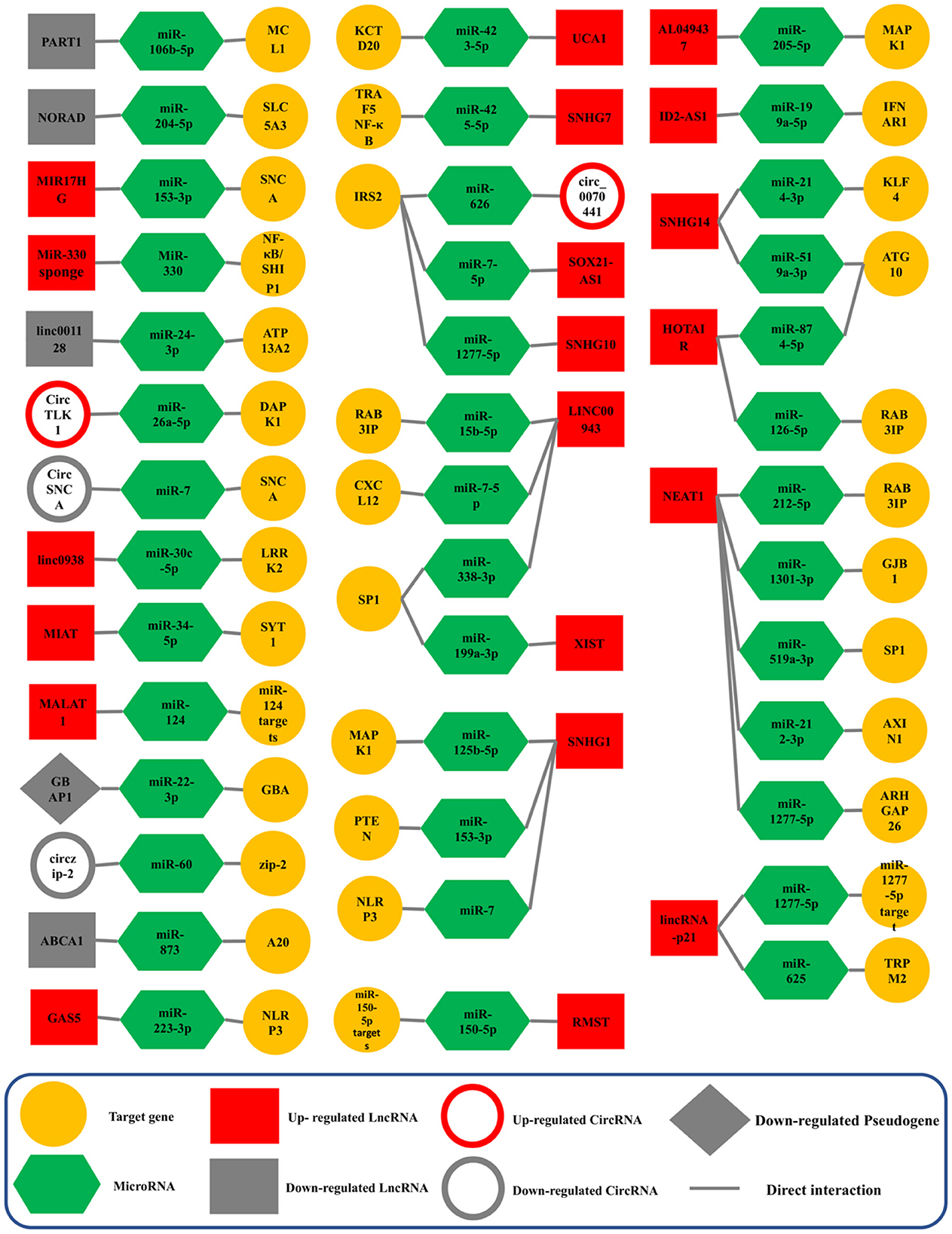 Competing endogenous RNA (ceRNA) networks in Parkinson’s disease: A systematic reviewMohammad Reza Asadi, Samin Abed, Ghazal Kouchakali, and 8 more authors2023
Competing endogenous RNA (ceRNA) networks in Parkinson’s disease: A systematic reviewMohammad Reza Asadi, Samin Abed, Ghazal Kouchakali, and 8 more authors2023Parkinson’s disease (PD) is a distinctive clinical syndrome with several causes and clinical manifestations. Aside from an infectious cause, PD is a rapidly developing neurological disorder with a global rise in frequency. Notably, improved knowledge of molecular pathways and the developing novel diagnostic methods may result in better therapy for PD patients. In this regard, the amount of research on ceRNA axes is rising, highlighting the importance of these axes in PD. CeRNAs are transcripts that cross-regulate one another via competition for shared microRNAs (miRNAs). These transcripts may be either coding RNAs (mRNAs) or non-coding RNAs (ncRNAs). This research used a systematic review to assess validated loops of ceRNA in PD. The Prisma guideline was used to conduct this systematic review, which entailed systematically examining the articles of seven databases. Out of 309 entries, forty articles met all criteria for inclusion and were summarized in the appropriate table. CeRNA axes have been described through one of the shared vital components of the axes, including lncRNAs such as NEAT1, SNHG family, HOTAIR, MALAT1, XIST, circRNAs, and lincRNAs. Understanding the multiple aspects of this regulatory structure may aid in elucidating the unknown causal causes of PD and providing innovative molecular therapeutic targets and medical fields.
- Int J Pediatr Otorhinolaryngol
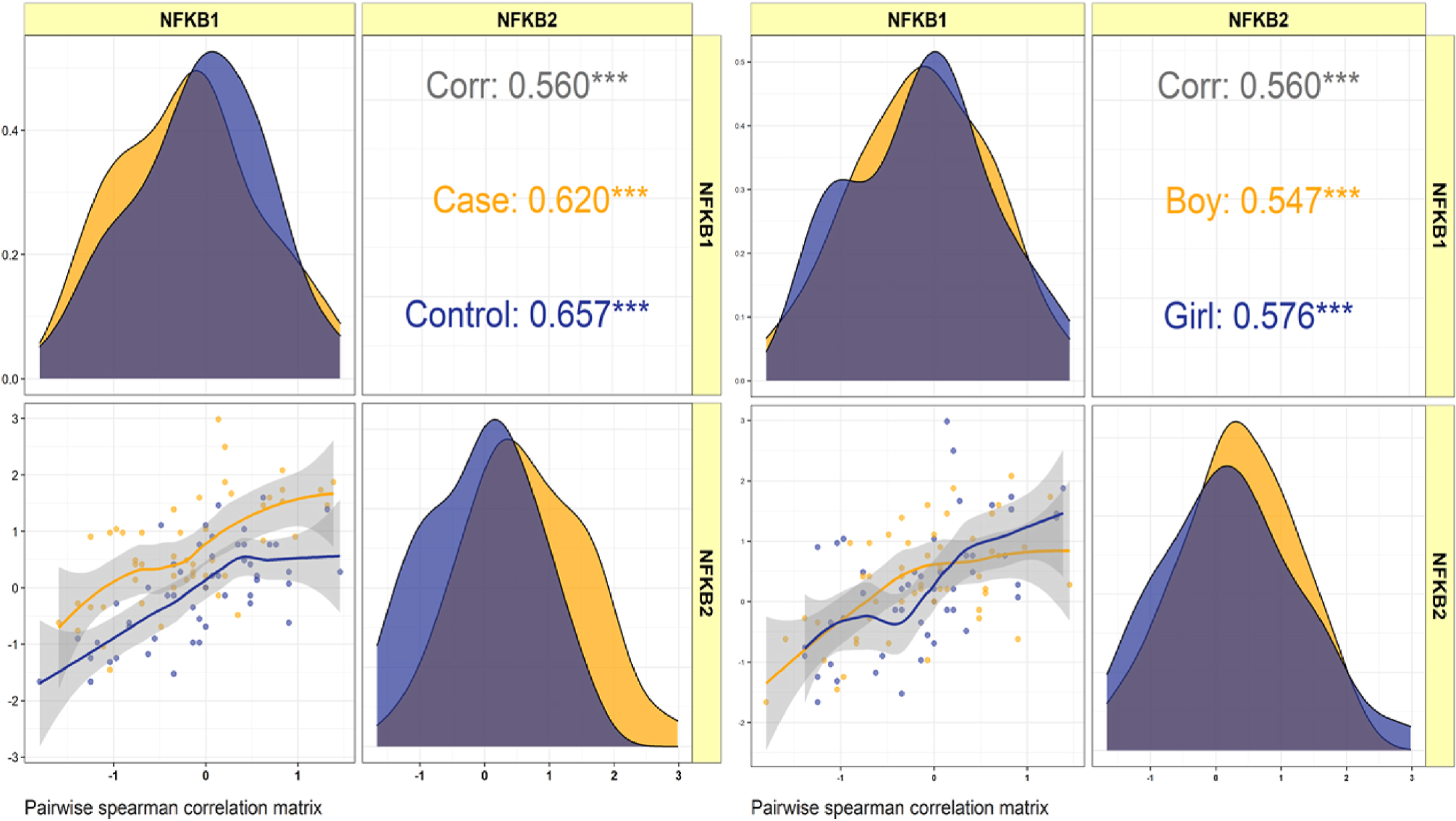 Analysis of NFKB1 and NFKB2 gene expression in the blood of patients with sudden sensorineural hearing lossYalda Jabbari Moghadam, Mohammad Reza Asadi, Vahdat Abbaszadeh, and 7 more authors2023
Analysis of NFKB1 and NFKB2 gene expression in the blood of patients with sudden sensorineural hearing lossYalda Jabbari Moghadam, Mohammad Reza Asadi, Vahdat Abbaszadeh, and 7 more authors2023Objectives Sudden Sensorineural Hearing Loss (SSNHL) is an increasingly common health problem today. Although the direct mortality rate of this disorder is relatively low, its impact on quality of life is enormous; this is why accurate identification of pathogenesis and influencing factors in the disease process can play an essential role in preventing and treating the disease. Acute inflammation, which leads to chronic inflammation due to aberrant expression of inflammation-mediating genes, may play a significant role in the pathogenesis of the disease. The essential Nuclear factor kappa B (NF-kB) pathway genes, NFKB1 and NFKB2, serve as prothrombotic agents when expressed abnormally, compromising the cochlea by disrupting the endolymphatic potential and causing SSNHL. Methods This study investigates the expression levels of NFKB1 and NFKB2 in peripheral blood (PB) through a quantitative polymerase chain reaction in 50 Iranian patients with SSNHL, and 50 healthy volunteers were of the same age and sex as controls. Results As a result, NFKB2 expression levels in patients were higher than in controls, regardless of sex or age (posterior beta = 0.619, adjusted P-value = 0.016), and NFKB1 expression levels did not show significant differences between patients and controls. The expression levels of NFKB1 and NFKB2 had significantly strong positive correlations in both SSNHL patients and healthy individuals (r = 0.620, P = 0.001 and r = 0.657, P 0.001, respectively), suggesting the presence of an interconnected network. Conclusion NFKB2 has been identified as a significant inflammatory factor in the pathophysiology of SSNHL disease. Inflammation can play an essential role in developing SSNHL, and our findings could be used as a guide for future research.
- Metab Brain Dis
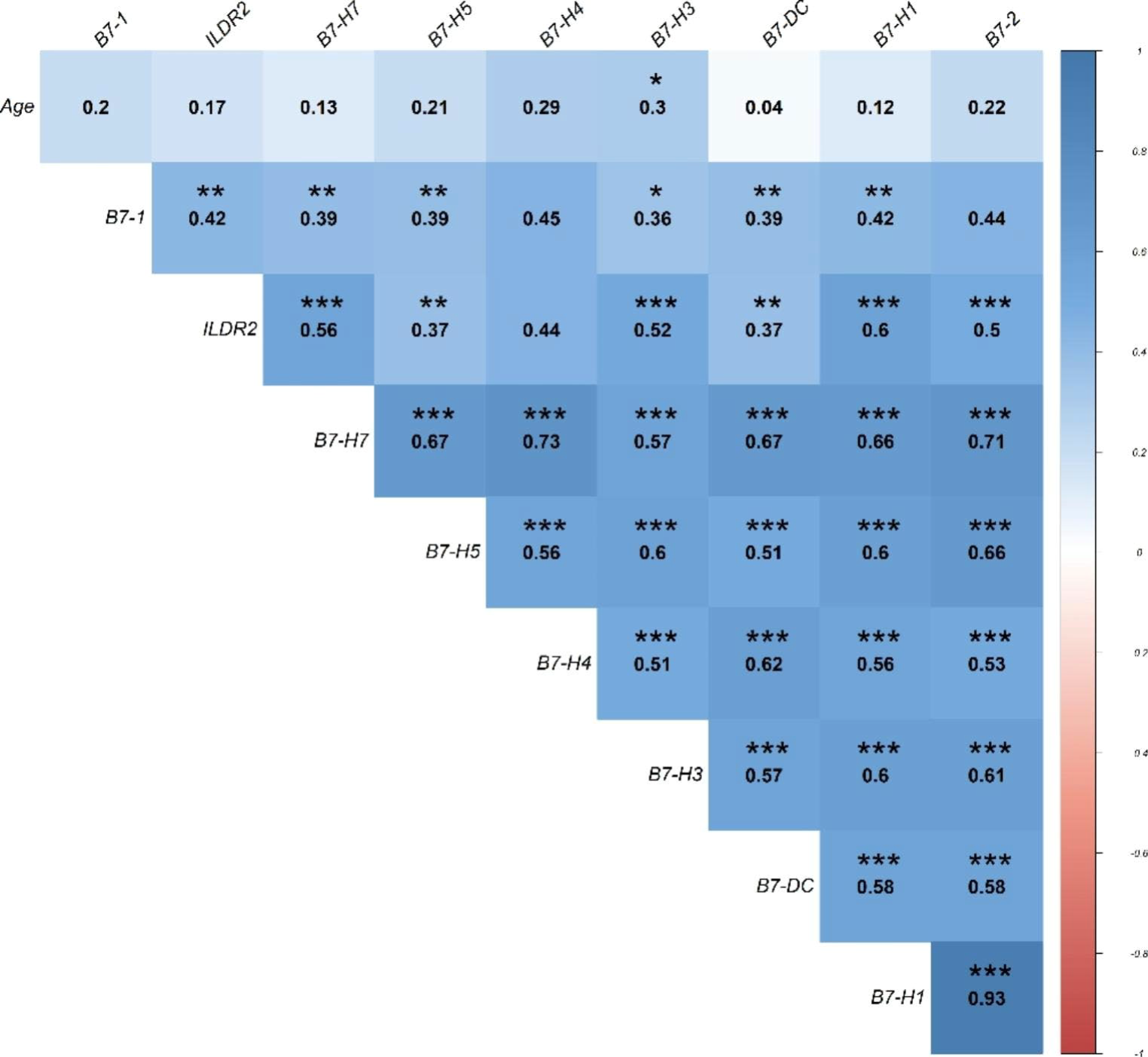 Expression analysis of inhibitory B7 family members in Alzheimer’s diseaseHani Sabaie, Parham Tamimi, Jalal Gharesouran, and 7 more authors2023
Expression analysis of inhibitory B7 family members in Alzheimer’s diseaseHani Sabaie, Parham Tamimi, Jalal Gharesouran, and 7 more authors2023Alzheimer’s disease (AD) is a global health problem due to its complexity, which frequently makes the development of treatment methods extremely difficult. Therefore, new methodologies are necessary to investigate the pathophysiology of AD and to treat AD. The interaction of immune modulation and neurodegeneration has added new dimensions in current knowledge of AD etiology and offers an attractive opportunity for the discovery of novel biomarkers and therapies. Using quantitative polymerase chain reaction, we compared the expression levels of inhibitory B7 family members (B7-1, B7-2, B7-H1, B7-DC, B7-H3, B7-H4, B7-H5, B7-H7, and ILDR2), as immune regulators, in the peripheral blood of late-onset AD (LOAD) patients (n = 50) and healthy individuals (n = 50). The levels of B7-2, B7-H4, ILDR2, and B7-DC expression were significantly higher in-patient blood samples than in control blood samples. Furthermore, we discovered a substantial positive correlation between all gene expression levels. In addition, the current study indicated that ILDR2, B7-H4, B7-2, and B7-DC might serve as diagnostic biomarkers to identify LOAD patients from healthy persons. The present work provides additional evidence for the significance of inhibitory B7 family members to the etiology of LOAD.
2022
- BMC Psychiatry
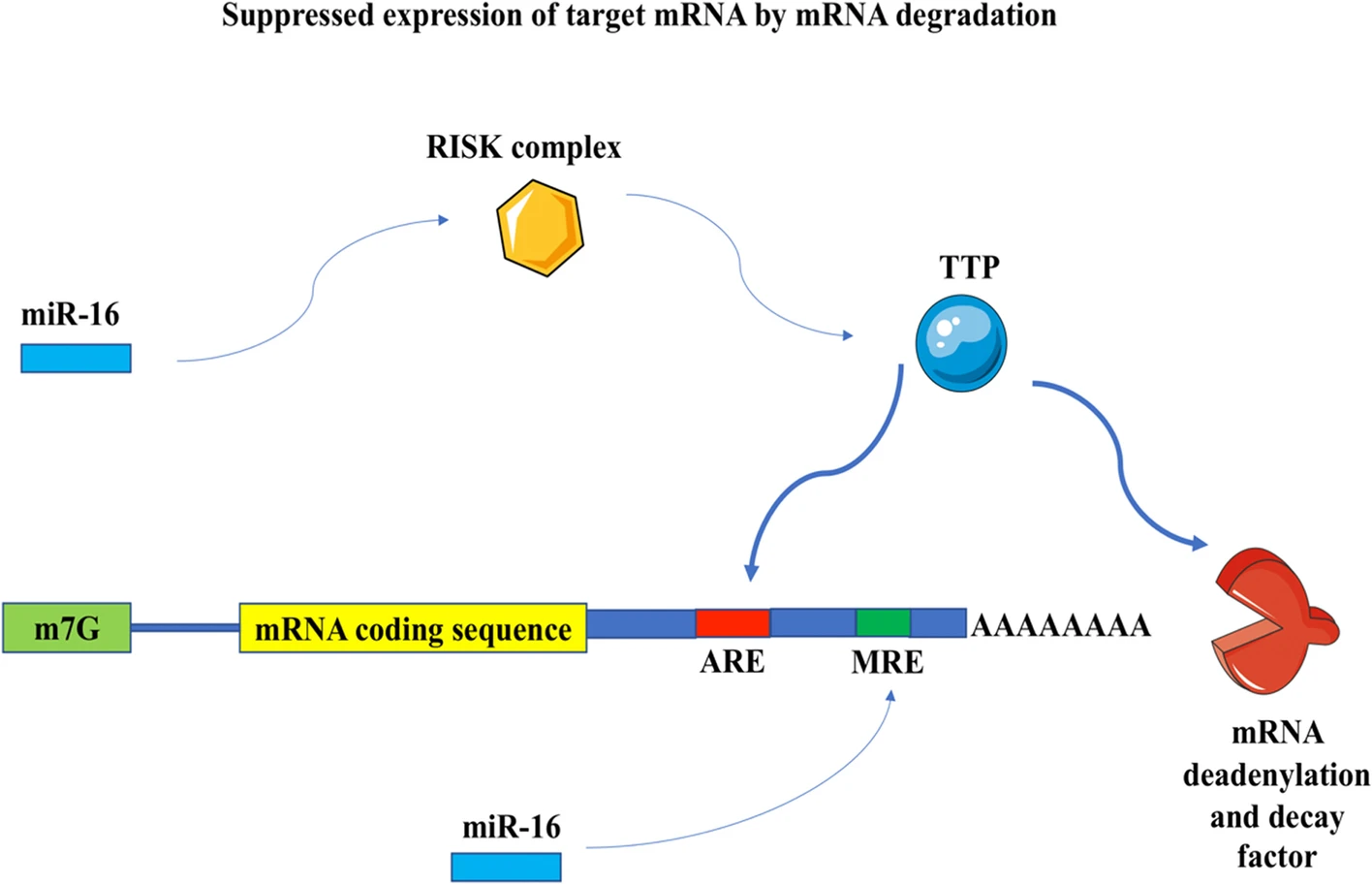 Assessing the expression of two post-transcriptional BDNF regulators, TTP and miR-16 in the peripheral blood of patients with SchizophreniaMohammad Reza Asadi, Jalal Gharesouran, Hani Sabaie, and 8 more authors2022
Assessing the expression of two post-transcriptional BDNF regulators, TTP and miR-16 in the peripheral blood of patients with SchizophreniaMohammad Reza Asadi, Jalal Gharesouran, Hani Sabaie, and 8 more authors2022Schizophrenia (SCZ) is a severe mental disorder with an unknown pathophysiology. Brain-Derived Neurotrophic Factor (BDNF) is a neurotrophin that has been associated with synapse plasticity, learning, and memory, as well as neurodevelopment and neuroprotection. The importance of neurodevelopmental and neurotoxicity-related components in the pathophysiology of SCZ has been highlighted in research on the neurobiology of this disease. The purpose of this research is to investigate the significant expression of two variables, tristetraprolin (TTP) and miR-16, which are known to be regulators of BDNF expression. Fifty Iranian Azeri SCZ patients were enrolled, and fifty healthy volunteers were age- and gender-matched as controls. A quantitative polymerase chain reaction measured the expression levels of the TTP and miR-16 in the peripheral blood (PB) of SCZ patients and healthy people. TTP expression levels in patients were higher than in controls, regardless of gender or age (posterior beta = 1.532, adjusted P-value = 0.012). TTP and miR-16 expression levels were found to be significantly correlated in both SCZ patients and healthy controls (r = 0.701, P < 0.001 and r = 0.777, P < 0.001, respectively). Due to the increased expression of TTP in SCZ and the existence of a significant correlation between TTP and miR-16, which helps to act on target mRNAs with AU-rich elements, this mechanism can be considered an influencing factor in SCZ.
- Front Oncol
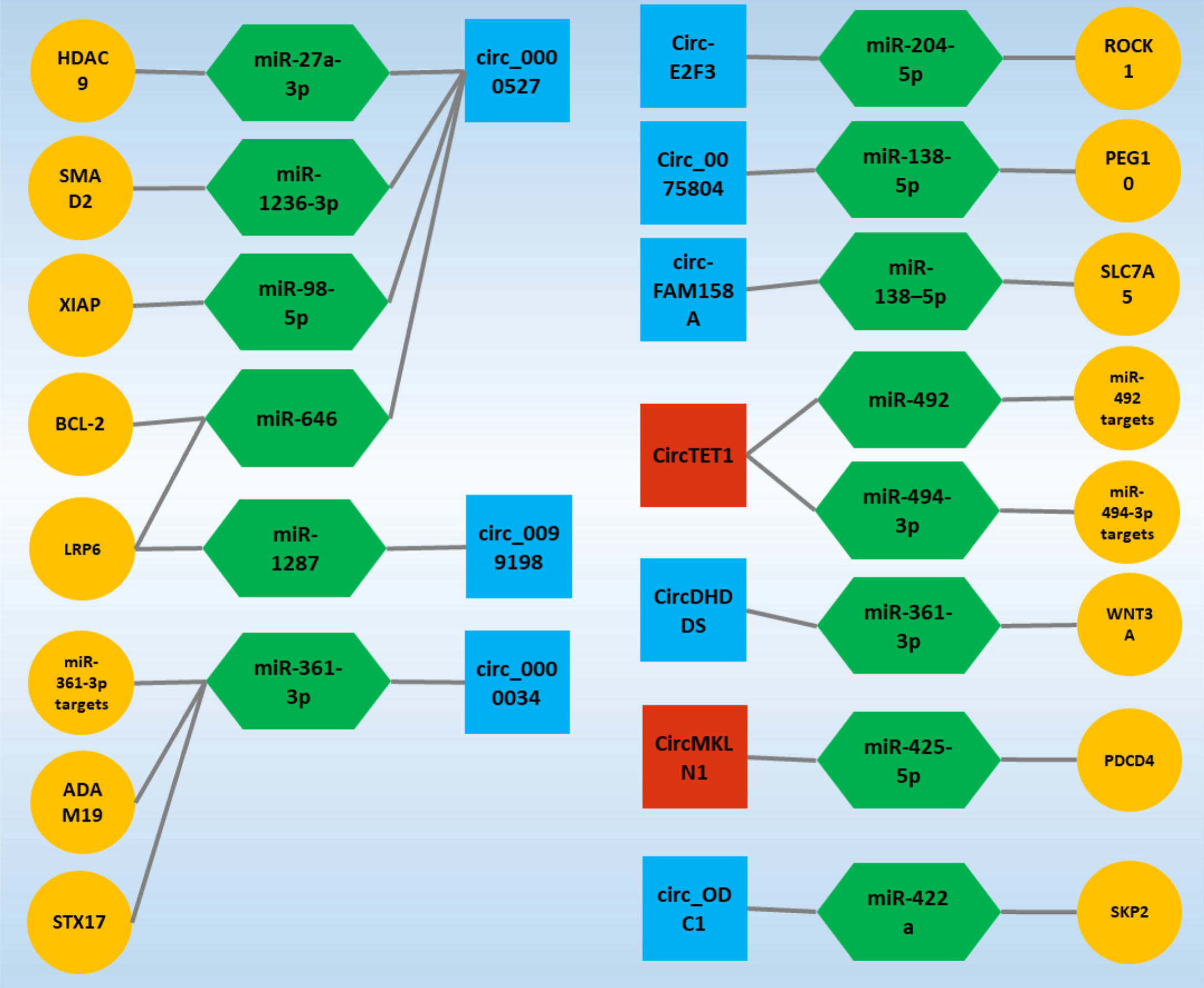 CircRNA-Associated CeRNAs Regulatory Axes in Retinoblastoma: A Systematic Scoping ReviewMohammad Reza Asadi, Marziyeh Sadat Moslehian, Hani Sabaie, and 6 more authors2022
CircRNA-Associated CeRNAs Regulatory Axes in Retinoblastoma: A Systematic Scoping ReviewMohammad Reza Asadi, Marziyeh Sadat Moslehian, Hani Sabaie, and 6 more authors2022Retinoblastoma (RB) is one of the most common childhood cancers caused by RB gene mutations (tumor suppressor gene in various patients). A better understanding of molecular pathways and the development of new diagnostic approaches may lead to better treatment for RB patients. The number of studies on ceRNA axes is increasing, emphasizing the significance of these axes in RB. Circular RNAs (circRNAs) play a vital role in competing endogenous RNA (ceRNA) regulatory axes by sponging microRNAs and regulating gene expression. Because of the broadness of ceRNA interaction networks, they may assist in investigating treatment targets in RB. This study conducted a systematic scoping review to evaluate verified loops of ceRNA in RB, focusing on the ceRNA axis and its relationship to circRNAs. This scoping review was carried out using a six-step strategy and the Prisma guideline, and it involved systematically searching the publications of seven databases. Out of 363 records, sixteen articles were entirely consistent with the defined inclusion criteria and were summarized in the relevant table. The majority of the studies focused on the circRNAs circ_0000527, circ_0000034, and circTET1, with approximately two-fifths of the studies focusing on a single circRNA. Understanding the many features of this regulatory structure may help elucidate RB’s unknown causative factors and provide novel molecular potential therapeutic targets and medical fields.
- Front Aging Neurosci
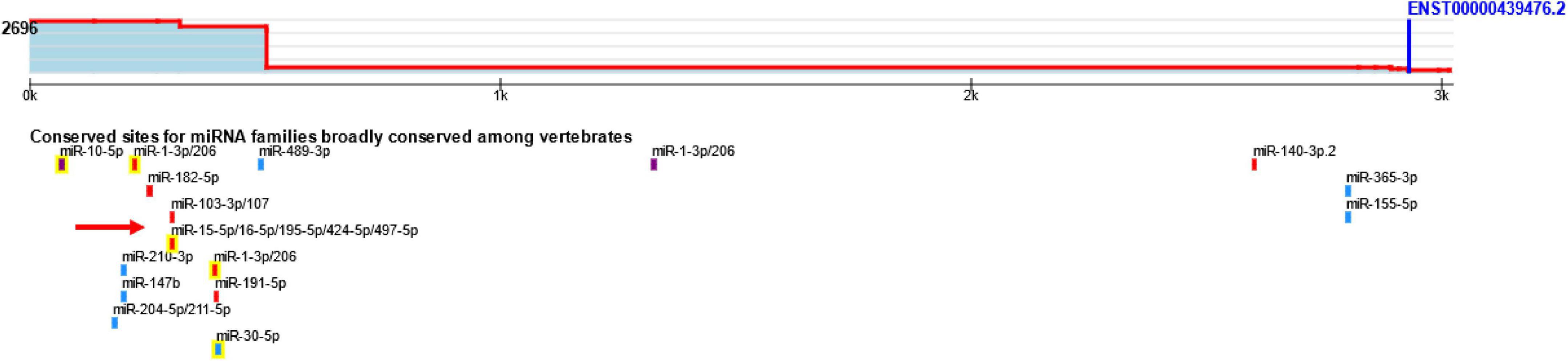 Analysis of ROQUIN, Tristetraprolin (TTP), and BDNF/miR-16/TTP regulatory axis in late onset Alzheimer’s diseaseMohammad Reza Asadi, Mahnaz Talebi, Jalal Gharesouran, and 6 more authors2022
Analysis of ROQUIN, Tristetraprolin (TTP), and BDNF/miR-16/TTP regulatory axis in late onset Alzheimer’s diseaseMohammad Reza Asadi, Mahnaz Talebi, Jalal Gharesouran, and 6 more authors2022Alzheimer’s disease (AD) is a heterogeneous degenerative disorder of the brain that is on the rise worldwide. One of the critical processes that might be disturbed in AD is gene expression regulation. Tristetraprolin (TTP) and RC3H1 gene (ROQUIN) are two RNA-binding proteins (RBPs) that target AU-rich elements (AREs) and constitutive decay elements (CDEs), respectively. TTP and ROQUIN, members of the CCCH zinc-finger protein family, have been demonstrated to fine-tune numerous inflammatory factors. In addition, miR-16 has distinct characteristics and may influence the target mRNA through the ARE site. Interestingly, BDNF mRNA has ARE sites in the 3’ untranslated region (UTR) and can be targeted by regulatory factors, such as TTP and miR-16 on MRE sequences, forming BDNF/miR-16/TTP regulatory axis. A number of two microarray datasets were downloaded, including information on mRNAs (GSE106241) and miRNAs (GSE157239) from individuals with AD and corresponding controls. R software was used to identify BDNF, TTP, ROQUIN, and miR-16 expression levels in temporal cortex (TC) tissue datasets. Q-PCR was also used to evaluate the expression of these regulatory factors and the expression of BDNF in the blood of 50 patients with AD and 50 controls. Bioinformatic evaluation showed that TTP and miR-16 overexpression might act as post-transcriptional regulatory factors to control BDNF expression in AD in TC samples. Instead, this expression pattern was not found in peripheral blood samples from patients with AD compared to normal controls. ROQUIN expression was increased in the peripheral blood of patients with AD. Hsa-miR-16-5p levels did not show significant differences in peripheral blood samples. Finally, it was shown that TTP and BDNF, based on evaluating the receiver operating characteristic (ROC), effectively identify patients with AD from healthy controls. This study could provide a new perspective on the molecular regulatory processes associated with AD pathogenic mechanisms linked to the BDNF growth factor, although further research is needed on the possible roles of these factors in AD.
- Human Gene
 Association of seven fundamental genetic polymorphisms in long noncoding RNA MALAT1, SOX2OT and H19 with recurrent miscarriage in Turkish-Azeri Iranian populationParvin Hakimi, Naser Lotfalizad, Leyla Pabarja, and 8 more authors2022
Association of seven fundamental genetic polymorphisms in long noncoding RNA MALAT1, SOX2OT and H19 with recurrent miscarriage in Turkish-Azeri Iranian populationParvin Hakimi, Naser Lotfalizad, Leyla Pabarja, and 8 more authors2022Background Recurrent miscarriage (RM), at a rate of 1% of all couples trying to conceive, is one of the main problems in pregnancy. The root cause of the disease has not been determined, but genetic factors and lifestyle influence the process. The minor change in the expression of lncRNAs shows its significant effects on various complications, one of which can be RM. SNPs are single nucleotide polymorphisms that have the potential to alter the function and expression of lncRNAs. Material and methods This study investigated seven SNPs in RM with 400 cases and 400 controls in the Iranian Turkish Azeri population using ARMS-PCR, including the MALAT1 lncRNA rs591291 rs3200401, rs619586, rs664589, and rs656605, SOX2OT lncRNA rs9839776, and H19 lncRNA rs3741216 variants. Results MALAT1 lncRNA rs591291 rs3200401, rs619586, rs664589, and rs656605, SOX2OT lncRNA rs9839776, and H19 lncRNA rs3741216 variants. We found that the MALAT1 lncRNA rs619586G variant in the co-dominant (AA vs. AG; OR = 0.65, 95% CI = 0.47–0.9 and AA vs. GG; OR = 0.5, 95% CI = 0.26–0.99, P = 0.0074), the dominant (AG + GG vs. AA; OR = 0.63, 95% CI = 0.46–0.85, P = 0.0023) and over-dominant (AA+GG vs. AG; OR = 0.68, 95% CI = 0.5–0.93, P = 0.016) inherited models has an association with decreased risk of RM and the MALAT1 lncRNA rs591291- rs3200401- rs619586 - rs664589 - rs656605 (C-C-G-C-G) haplotype is most likely a haplotype prone to RM. Conclusion In conclusion, our findings propose that the rs619586 G variant in the Iranian Turkish Azeri may have potential protective effects, lowering the risk of RM.
- Exp Mol Pathol
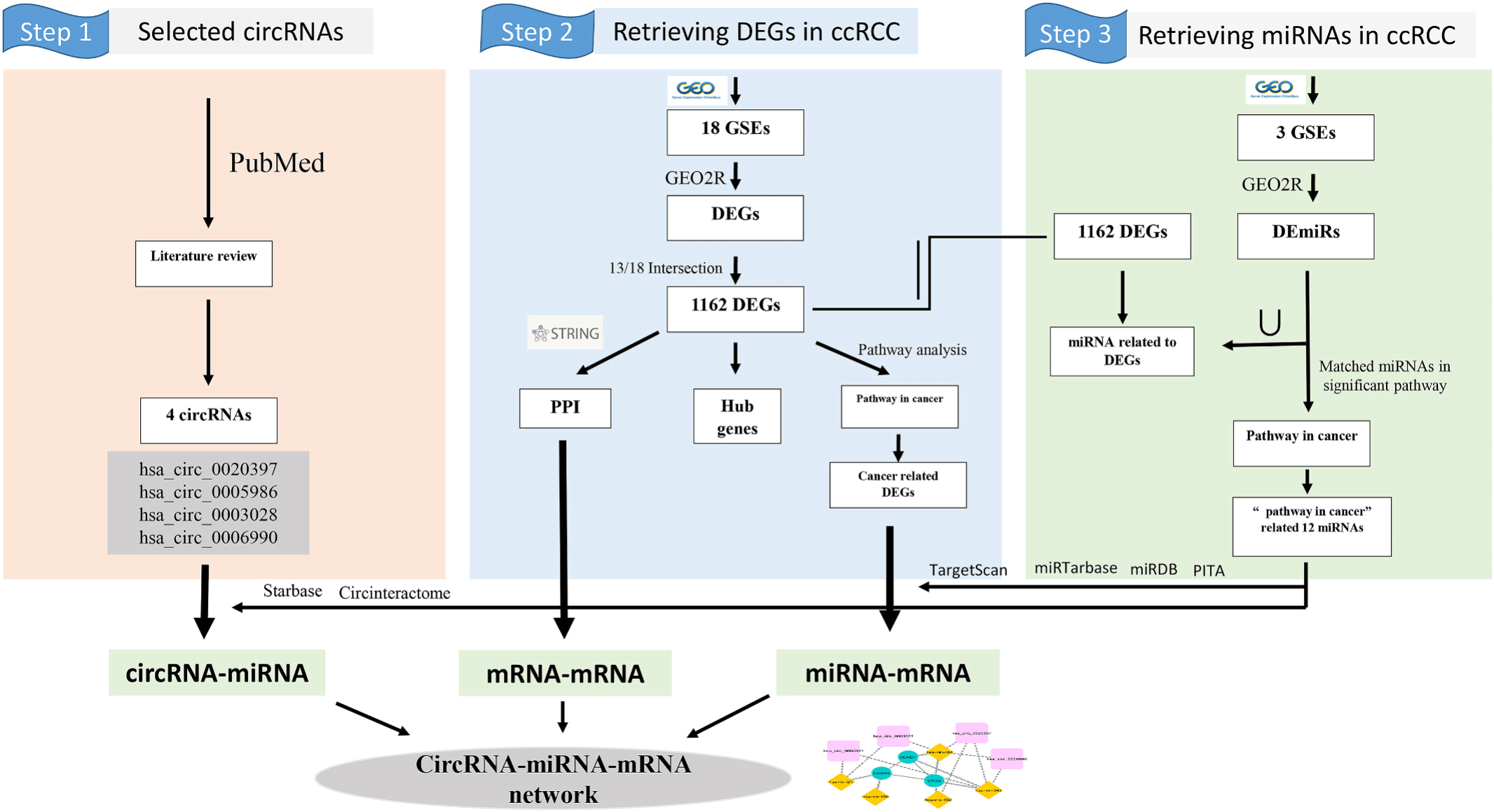 Expression analysis of hsa_circ_0020397, hsa_circ_0005986, hsa_circ_0003028, and hsa_circ_0006990 in renal cell carcinomaElham Mohammadisoleimani, Zahra Firoozi, Mohammad Mehdi Naghizadeh, and 9 more authors2022
Expression analysis of hsa_circ_0020397, hsa_circ_0005986, hsa_circ_0003028, and hsa_circ_0006990 in renal cell carcinomaElham Mohammadisoleimani, Zahra Firoozi, Mohammad Mehdi Naghizadeh, and 9 more authors2022Renal cell carcinoma (RCC) is a prevalent heterogeneous kidney cancer. So far, different genes have been reported for RCC development. However, its particular molecular mechanism remains unclear. Circular RNAs (circRNAs), a class of non-coding RNAs, are involved in numerous biological processes in different malignancies such as RCC. This study aims to assess the expression and underlying mechanism of four circRNAs (hsa_circ_0020397, hsa_circ_0005986, hsa_circ_0003028, hsa_circ_0006990) with possible new roles in RCC. In the experimental step, we investigated the expression of these four circRNAs in our RCC samples using quantitative real-time polymerase chain reaction. In the bioinformatics step, the differential expressed mRNAs (DEmRNAs), and miRNAs (DEmiRNAs) were obtained from the GEO datasets using the GEO2R tool. A protein-protein interaction network was constructed using the STRING database, and hub genes were identified by Cytoscape. Molecular pathways associated with hub genes were detected using KEGG pathway enrichment analysis. Then, we utilized the ToppGene database to detect the relationships between DEmiRNAs and hub genes. Furthermore, interactions between circRNAs and DEmiRNAs were predicted by the StarBase and circinteractome databases. Finally, a circRNA-DEmiRNA-hub gene triple network was constructed. Our results revealed that the expression of hsa_circ_0020397, hsa_circ_0005986, and hsa_circ_0006990 was downregulated in RCC tissues. Moreover, these circRNAs had a significantly lower expression in patients with a history of kidney disease. Furthermore, hsa_circ_0003028 and hsa_circ_0006990 showed higher expression in the tumor of participants with Lymphovascular/perineural invasion and oncocytoma type, respectively. Based on bioinformatic results, 15 circRNA-DEmiRNA-hub gene ceRNA regulatory axes were predicted, which included three hub genes, five miRNAs, and four selected circRNAs. In conclusion, the current work is the first to emphasize the expression of the hsa_circ_0020397, hsa_circ_0005986, hsa_circ_0003028, and hsa_circ_0006990 in RCC patients presents a novel perspective on the molecular processes underlying the pathogenic mechanisms of RCC.
- Metab Brain Dis
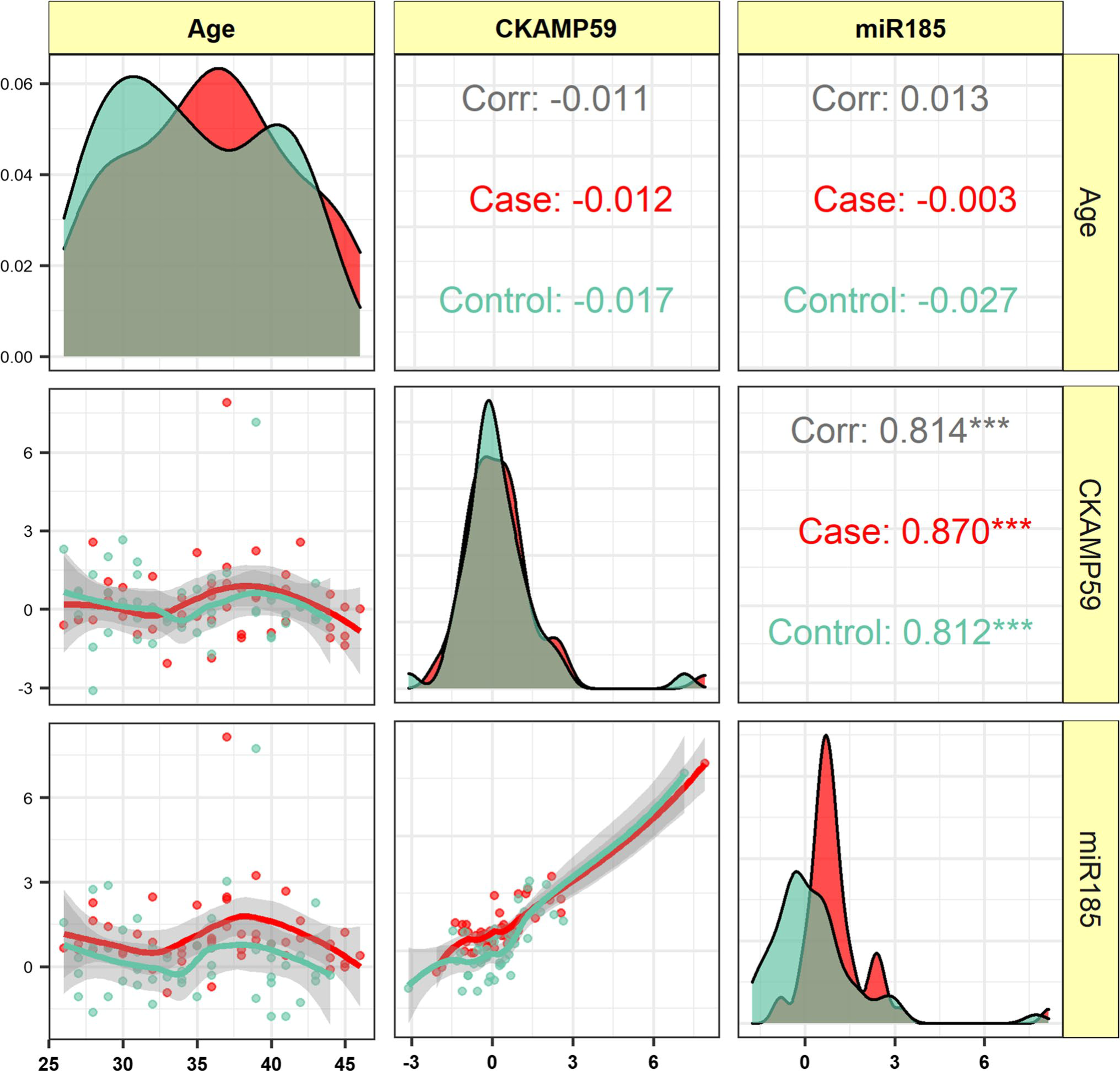 Downregulation of miR-185 is a common pathogenic event in 22q11.2 deletion syndrome-related and idiopathic schizophreniaHani Sabaie, Jalal Gharesouran, Mohammad Reza Asadi, and 7 more authors2022
Downregulation of miR-185 is a common pathogenic event in 22q11.2 deletion syndrome-related and idiopathic schizophreniaHani Sabaie, Jalal Gharesouran, Mohammad Reza Asadi, and 7 more authors2022Schizophrenia (SCZ) is known as a complicated mental disease with an unknown etiology. The microdeletion of 22q11.2 is the most potent genetic risk factor. Researchers are still trying to find which genes in the deletion region are linked to SCZ. MIR185, encoding microRNA (miR)-185, is present in the minimal 1.5 megabase deletion. Nonetheless, the miR-185 expression profile and its corresponding target genes in animal models and patients with 22q11.2 deletion syndrome (22q11.2DS) imply that more study is required about miR-185 and its corresponding downstream pathways within idiopathic SCZ. The expression of hsa-miR-185-5p and its corresponding target gene, shisa family member 7 (SHISA7), sometimes called CKAMP59, were evaluated in the peripheral blood (PB) samples of Iranian Azeri patients with idiopathic SCZ and healthy subjects, matched by gender and age as control groups by quantitative polymerase chain reaction (qPCR). Fifty SCZ patients (male/female: 22/28, age (mean ± standard deviation (SD)): 35.9 ± 5.6) and 50 matched healthy controls (male/female: 23/27, age (mean ± SD): 34.7 ± 5.4) were enrolled. The expression of hsa-miR-185-5p in the PB samples from subjects with idiopathic SCZ was substantially lower than in that of control groups (posterior beta = -0.985, adjusted P-value < 0.0001). There was also a difference within the expression profile between female and male subgroups (posterior beta = -0.86, adjusted P-value = 0.046 and posterior beta = -1.015, adjusted P-value = 0.004, in turn). Nevertheless, no significant difference was present in the expression level of CKAMP59 between PB samples from patients and control groups (adjusted P-value > 0.999). The analysis of the receiver operating characteristic (ROC) curve suggested that hsa-miR-185-5p may correctly distinguish subjects with idiopathic SCZ from healthy people (the area under curve (AUC) value: 0.722). Furthermore, there was a strong positive correlation between the expression pattern of the abovementioned genes in patients with SCZ and healthy subjects (r = 0.870, P < 0.001 and r = 0.812, P < 0.001, respectively), indicating that this miR works as an enhancer. More research is needed to determine if the hsa-miR-185-5p has an enhancer activity. In summary, this is the first research to highlight the expression of the miR-185 and CKAMP59 genes in the PB from subjects with idiopathic SCZ. Our findings suggest that gene expression alterations mediated by miR-185 may play a role in the pathogenesis of idiopathic and 22q11.2DS SCZ. It is worth noting that, despite a substantial and clear relationship between CKAMP59 and hsa-miR-185-5p, indicating an interactive network, their involvement in the development of SCZ should be reconsidered based on the whole blood sample since the changed expression level of CKAMP59 was not significant. Further research with greater sample sizes and particular leukocyte subsets can greatly make these results stronger.
- Front Psychiatry
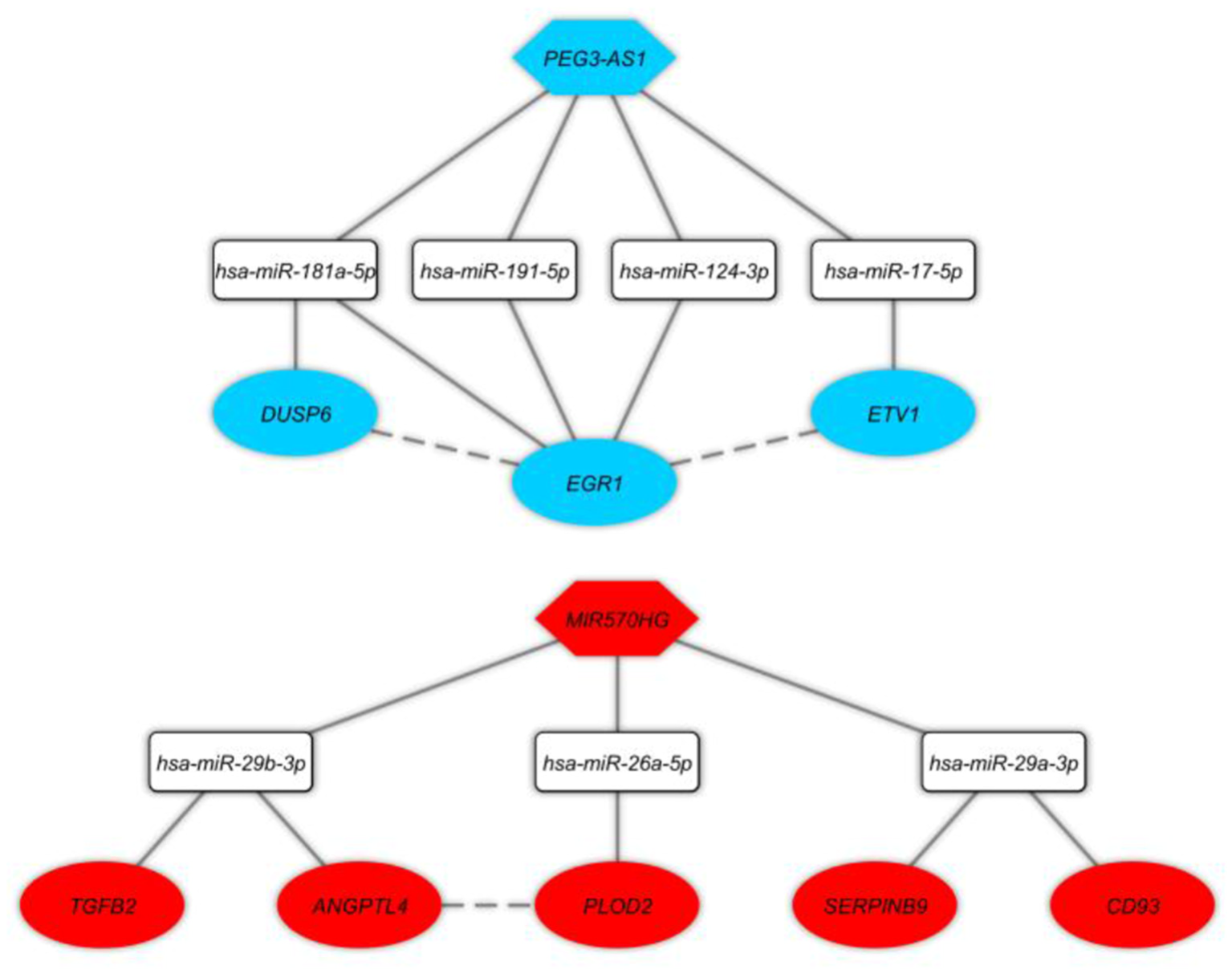 Identification of key long non-coding RNA-associated competing endogenous RNA axes in Brodmann Area 10 brain region of schizophrenia patientsHani Sabaie, Mahdi Gholipour, Mohammad Reza Asadi, and 7 more authors2022
Identification of key long non-coding RNA-associated competing endogenous RNA axes in Brodmann Area 10 brain region of schizophrenia patientsHani Sabaie, Mahdi Gholipour, Mohammad Reza Asadi, and 7 more authors2022Schizophrenia (SCZ) is a serious mental condition with an unknown cause. According to the reports, Brodmann Area 10 (BA10) is linked to the pathology and cortical dysfunction of SCZ, which demonstrates a number of replicated findings related to research on SCZ and the dysfunction in tasks requiring cognitive control in particular. Genetics’ role in the pathophysiology of SCZ is still unclear. Therefore, it may be helpful to understand the effects of these changes on the onset and progression of SCZ to find novel mechanisms involved in the regulation of gene transcription. In order to determine the molecular regulatory mechanisms affecting the SCZ, the long non-coding RNA (lncRNA)-associated competing endogenous RNAs (ceRNAs) axes in the BA10 area were determined using a bioinformatics approach in the present work. A microarray dataset (GSE17612) consisted of brain post-mortem tissues of the BA10 area from SCZ patients and matched healthy subjects was downloaded from the Gene Expression Omnibus (GEO) database. This dataset included probes for both lncRNAs and mRNAs. Using the R software’s limma package, the differentially expressed lncRNAs (DElncRNAs) and mRNAs (DEmRNAs) were found. The RNA interactions were also discovered using the DIANA-LncBase and miRTarBase databases. In the ceRNA network, positive correlations between DEmRNAs and DElncRNAs were evaluated using the Pearson correlation coefficient. Finally, lncRNA-associated ceRNA axes were built by using the co-expression and DElncRNA-miRNA-DEmRNA connections. We identified the DElncRNA-miRNA-DEmRNA axes, which included two key lncRNAs (PEG3-AS1, MIR570HG), seven key miRNAs (hsa-miR-124-3p, hsa-miR-17-5p, hsa-miR-181a-5p, hsa-miR-191-5p, hsa-miR-26a-5p, hsa-miR-29a-3p, hsa-miR-29b-3p), and eight key mRNAs (EGR1, ETV1, DUSP6, PLOD2, CD93, SERPINB9, ANGPTL4, TGFB2). Furthermore, DEmRNAs were found to be enriched in the “AGE-RAGE signaling pathway in diabetic complications”, “Amoebiasis”, “Transcriptional misregulation in cancer”, “Human T-cell leukemia virus 1 infection”, and “MAPK signaling pathway”. This study offers research targets for examining significant molecular pathways connected to the pathogenesis of SCZ, even though the function of these ceRNA axes still needs to be investigated.
- Front Genet
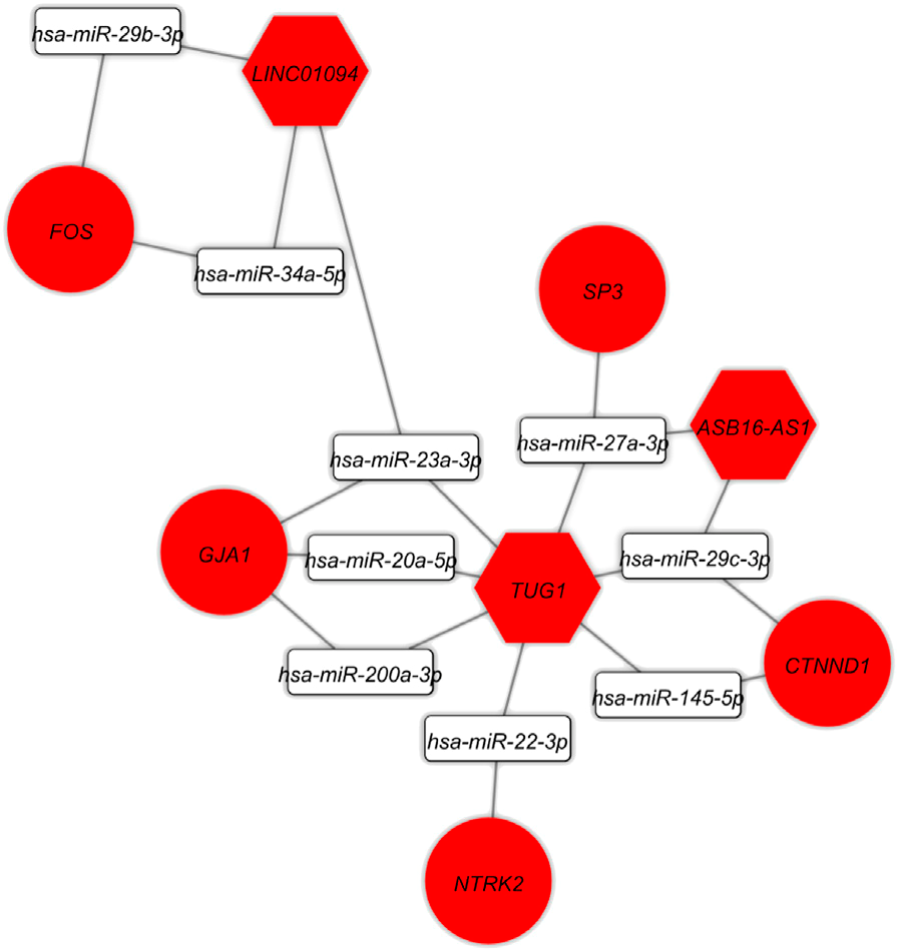 Identification of potential regulatory long non-coding RNA-associated competing endogenous RNA axes in periplaque regions in multiple sclerosisHani Sabaie, Sharareh Khorami Rouz, Ghazal Kouchakali, and 7 more authors2022
Identification of potential regulatory long non-coding RNA-associated competing endogenous RNA axes in periplaque regions in multiple sclerosisHani Sabaie, Sharareh Khorami Rouz, Ghazal Kouchakali, and 7 more authors2022Slow-burning inflammation at the lesion rim is connected to the expansion of chronic multiple sclerosis (MS) lesions. However, the underlying processes causing expansion are not clearly realized. In this context, the current study used a bioinformatics approach to identify the expression profiles and related lncRNA-associated ceRNA regulatory axes in the periplaque region in MS patients. Expression data (GSE52139) from periplaque regions in the secondary progressive MS spinal cord and controls were downloaded from the Gene Expression Omnibus database (GEO), which has details on mRNAs and lncRNAs. Using the R software’s limma package, the differentially expressed lncRNAs (DElncRNAs) and mRNAs (DEmRNAs) were found. The RNA interactions were also found using the DIANA-LncBase, miRTarBase, and HMDD databases. The Pearson correlation coefficient was used to determine whether there were any positive correlations between DEmRNAs and DElncRNAs in the ceRNA network. Finally, lncRNA-associated ceRNA axes were created based on co-expression and connections between DElncRNA, miRNA, and DEmRNA. We used the Enrichr tool to enrich the biological process, molecular function, and pathways for DEmRNAs and DElncRNAs. A network of DEmRNAs’ protein-protein interactions was developed, and the top five hub genes were found using Cytoscape and STRING. The current study indicates that 15 DEmRNAs, including FOS, GJA1, NTRK2, CTNND1, and SP3, are connected to the MS ceRNA network. Additionally, four DElncRNAs (such as TUG1, ASB16-AS1, and LINC01094) that regulated the aforementioned mRNAs by sponging 14 MS-related miRNAs (e.g., hsa-miR-145-5p, hsa-miR-200a-3p, hsa-miR-20a-5p, hsa-miR-22-3p, hsa-miR-23a-3p, hsa-miR-27a-3p, hsa-miR-29b-3p, hsa-miR-29c-3p, hsa-miR-34a-5p) were found. In addition, the analysis of pathway enrichment revealed that DEmRNAs were enriched in the pathways for the “MAPK signaling pathway”, “Kaposi sarcoma-associated herpesvirus infection”, “Human immunodeficiency virus one infection”, “Lipid and atherosclerosis”, and “Amphetamine addiction”. Even though the function of these ceRNA axes needs to be investigated further, this study provides research targets for studying ceRNA-mediated molecular mechanisms related to periplaque demyelination in MS.
- Front Aging Neurosci
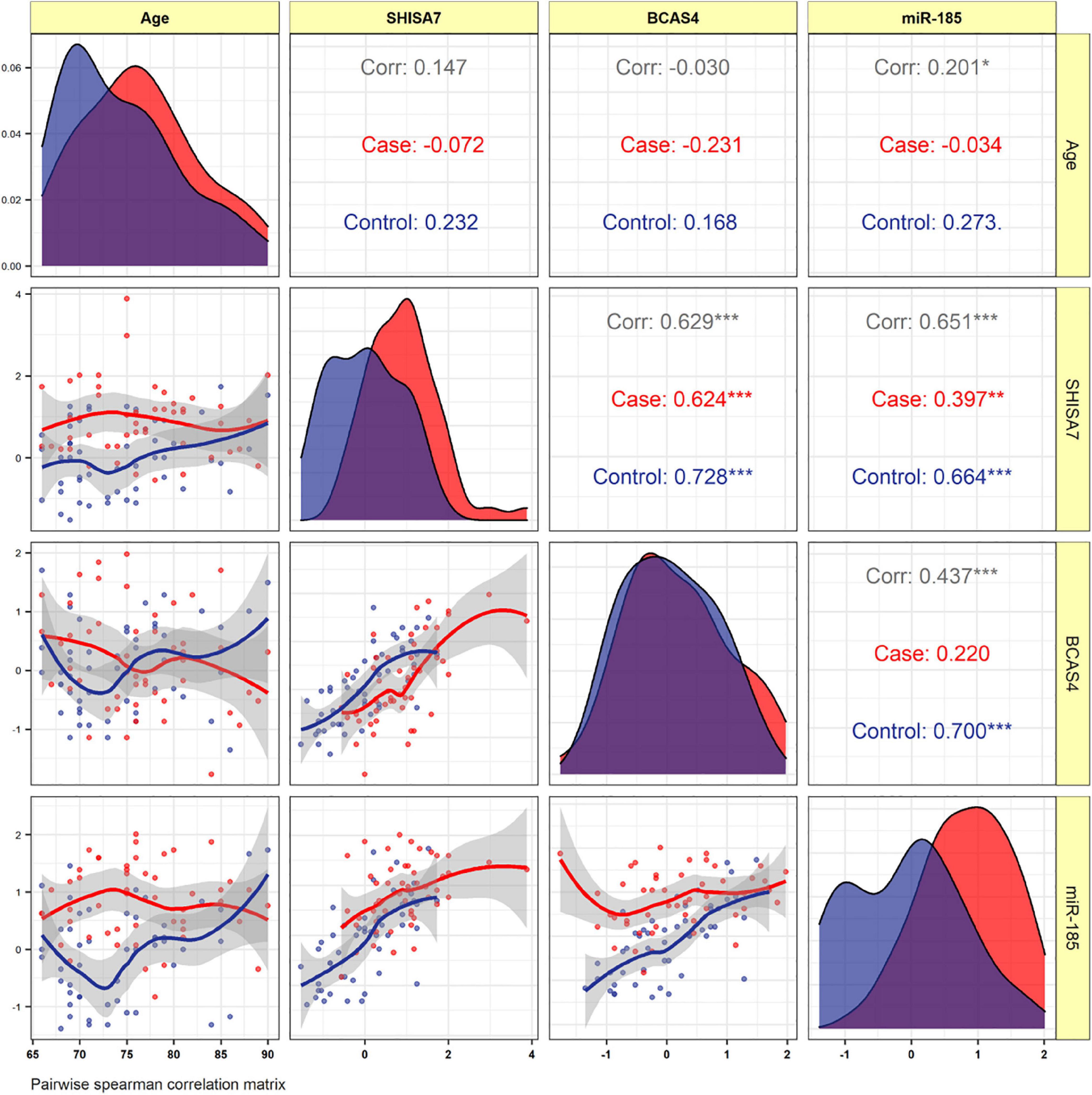 Identification and Analysis of BCAS4/hsa-miR-185-5p/SHISA7 Competing Endogenous RNA Axis in Late-Onset Alzheimer’s Disease Using Bioinformatic and Experimental ApproachesHani Sabaie, Mahnaz Talebi, Jalal Gharesouarn, and 7 more authors2022
Identification and Analysis of BCAS4/hsa-miR-185-5p/SHISA7 Competing Endogenous RNA Axis in Late-Onset Alzheimer’s Disease Using Bioinformatic and Experimental ApproachesHani Sabaie, Mahnaz Talebi, Jalal Gharesouarn, and 7 more authors2022Alzheimer’s disease (AD) is a heterogeneous degenerative brain disorder with a rising prevalence worldwide. SHISA7 (CKAMP59) has emerged as one of the most intriguing new members of the SHISA family, in that, unlike other CKAMP counterparts, it exhibits a direct function in inhibitory synaptic GABAAR regulation. We used bioinformatics and experimental methods in this research to explore competing endogenous RNA (ceRNA) regulation of BCAS4 and SHISA7 in tau pathogenesis and their capacity as peripheral biomarkers linked to an abnormal inflammatory response in AD. The Gene Expression Omnibus database included two microarray datasets, including information on mRNAs (GSE106241) and miRNAs (GSE157239) from individuals with AD with different degrees of AD-associated neurofibrillary pathology in the temporal cortex (TC) tissue specimens and corresponding controls were downloaded from the Gene Expression Omnibus database. The limma package in the R software was used to identify differently expressed mRNAs (DEmRNAs) and miRNAs (DEmiRNAs) associated with AD-related neurofibrillary pathology. Additionally, we used the quantitative polymerase chain reaction technique to examine the expression of the BCAS4/hsa-miR-185-5p/SHISA7 ceRNA axis in the peripheral blood (PB) of fifty AD patients and fifty control subjects. BCAS4 was shown to act as a ceRNA to control the SHISA7 expression throughout AD-associated neurofibrillary pathology in TC tissue specimens by sponging hsa-miR-185-5p, based on our bioinformatics study. Furthermore, in PB specimens from individuals suffering from AD and normal controls, we found no substantial differences in BCAS4 expression patterns. SHISA7 expression in AD patients’ PB was found to be reduced, as was the case in the TC. On the other hand, we discovered reduced amounts of hsa-miR-185-5p in AD patients’ PB samples compared to control subjects, unlike in TC tissue, where it had been demonstrated to be overexpressed. BCAS4 and SHISA7 expression levels showed a strong positive correlation, suggesting the presence of an interconnected network, most likely as a result of ceRNA regulation among PB specimens. The present study is the first evidence to highlight the expression of the BCAS4/miR-185-5p/SHISA7 ceRNA axis in the brain and PB of AD patients, and offers a new viewpoint on molecular processes underlying AD pathogenic mechanisms.
2021
- Int J Mol Sci
 The Regulatory Cross-Talk between microRNAs and Novel Members of the B7 Family in Human Diseases: A Scoping ReviewNoora Karim Ahangar, Nima Hemmat, Mohammad Khalaj-Kondori, and 9 more authors2021
The Regulatory Cross-Talk between microRNAs and Novel Members of the B7 Family in Human Diseases: A Scoping ReviewNoora Karim Ahangar, Nima Hemmat, Mohammad Khalaj-Kondori, and 9 more authors2021The members of the B7 family, as immune checkpoint molecules, can substantially regulate immune responses. Since microRNAs (miRs) can regulate gene expression post-transcriptionally, we conducted a scoping review to summarize and discuss the regulatory cross-talk between miRs and new B7 family immune checkpoint molecules, i.e., B7-H3, B7-H4, B7-H5, butyrophilin like 2 (BTNL2), B7-H6, B7-H7, and immunoglobulin like domain containing receptor 2 (ILDR2). The current study was performed using a six-stage methodology structure and Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guideline. PubMed, Embase, Scopus, Cochrane, ProQuest, and Google Scholar were systematically searched to obtain the relevant records to 5 November 2020. Two authors independently reviewed the obtained records and extracted the desired data. After quantitative and qualitative analyses, we used bioinformatics approaches to extend our knowledge about the regulatory cross-talk between miRs and the abovementioned B7 family members. Twenty-seven articles were identified that fulfilled the inclusion criteria. Studies with different designs reported gene–miR regulatory axes in various cancer and non-cancer diseases. The regulatory cross-talk between the aforementioned B7 family molecules and miRs might provide valuable insights into the pathogenesis of various human diseases.
- Front Aging Neurosci
 The Perspective of Dysregulated LncRNAs in Alzheimer’s Disease: A Systematic Scoping ReviewMohammad Reza Asadi, Mehdi Hassani, Shiva Kiani, and 6 more authors2021
The Perspective of Dysregulated LncRNAs in Alzheimer’s Disease: A Systematic Scoping ReviewMohammad Reza Asadi, Mehdi Hassani, Shiva Kiani, and 6 more authors2021LncRNAs act as part of non-coding RNAs at high levels of complex and stimulatory configurations in basic molecular mechanisms. Their extensive regulatory activity in the CNS continues on a small scale, from the functions of synapses to large-scale neurodevelopment and cognitive functions, aging, and can be seen in both health and disease situations. One of the vast consequences of the pathological role of dysregulated lncRNAs in the CNS due to their role in a network of regulatory pathways can be manifested in Alzheimer’s as a neurodegenerative disease. The disease is characterized by two main hallmarks: amyloid plaques due to the accumulation of β-amyloid components and neurofibrillary tangles (NFT) resulting from the accumulation of phosphorylated tau. Numerous studies in humans, animal models, and various cell lines have revealed the role of lncRNAs in the pathogenesis of Alzheimer’s disease. This scoping review was performed with a six-step strategy and based on the Prisma guideline by systematically searching the publications of seven databases. Out of 1,591 records, 69 articles were utterly aligned with the specified inclusion criteria and were summarized in the relevant table. Most of the studies were devoted to BACE1-AS, NEAT1, MALAT1, and SNHG1 lncRNAs, respectively, and about one-third of the studies investigated a unique lncRNA. About 56% of the studies reported up-regulation, and 7% reported down-regulation of lncRNAs expressions. Overall, this study was conducted to investigate the association between lncRNAs and Alzheimer’s disease to make a reputable source for further studies and find more molecular therapeutic goals for this disease.
- Front Aging Neurosci
 Stress Granules and Neurodegenerative Disorders: A Scoping ReviewMohammad Reza Asadi, Marziyeh Sadat Moslehian, Hani Sabaie, and 4 more authors2021
Stress Granules and Neurodegenerative Disorders: A Scoping ReviewMohammad Reza Asadi, Marziyeh Sadat Moslehian, Hani Sabaie, and 4 more authors2021Cytoplasmic ribonucleoproteins called stress granules (SGs) are considered as one of the main cellular solutions against stress. Their temporary presence ends with stress relief. Any factor such as chronic stress or mutations in the structure of the components of SGs that lead to their permanent presence can affect their interactions with pathological aggregations and increase the degenerative effects. SGs involved in RNA mechanisms are important factors in the pathophysiology of neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), frontotemporal degeneration (FTD), and Alzheimer’s diseases (AD). Although many studies have been performed in the field of SGs and neurodegenerative disorders, so far, no systematic studies have been executed in this field. The purpose of this study is to provide a comprehensive perspective of all studies about the role of SGs in the pathogenesis of neurodegenerative disorders with a focus on the protein ingredients of these granules. This scoping review is based on a six-stage methodology structure and the PRISMA guideline. A systematic search of seven databases for qualified articles was conducted until December 2020. Publications were screened independently by two reviewers and quantitative and qualitative analysis was performed on the extracted data. Bioinformatics analysis was used to plot the network and predict interprotein interactions. In addition, GO analysis was performed. A total of 48 articles were identified that comply the inclusion criteria. Most studies on neurodegenerative diseases have been conducted on ALS, AD, and FTD using human post mortem tissues. Human derived cell line studies have been used only in ALS. A total 29 genes of protein components of SGs have been studied, the most important of which are TDP-43, TIA-1, PABP-1. Bioinformatics studies have predicted 15 proteins to interact with the protein components of SGs, which may be the constituents of SGs. Understanding the interactions between SGs and pathological aggregations in neurodegenerative diseases can provide new targets for treatment of these disorders.
- Front Oncol
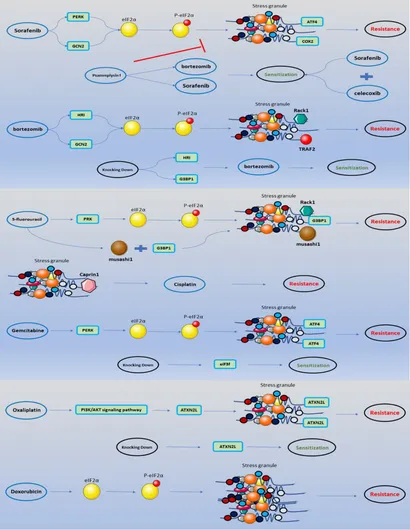 Stress Granules in the Anti-Cancer Medications Mechanism of Action: A Systematic Scoping ReviewMohammad Reza Asadi, Marziyeh Sadat Moslehian, Hani Sabaie, and 6 more authors2021
Stress Granules in the Anti-Cancer Medications Mechanism of Action: A Systematic Scoping ReviewMohammad Reza Asadi, Marziyeh Sadat Moslehian, Hani Sabaie, and 6 more authors2021Stress granule (SG) formation is a well-known cellular mechanism for minimizing stress-related damage and increasing cell survival. In addition to playing a critical role in the stress response, SGs have emerged as critical mediators in human health. It seems logical that SGs play a key role in cancer cell formation, development, and metastasis. Recent studies have shown that many SG components contribute to the anti-cancer medications’ responses through tumor-associated signaling pathways and other mechanisms. SG proteins are known for their involvement in the translation process, control of mRNA stability, and capacity to function in both the cytoplasm and nucleus. The current systematic review aimed to include all research on the impact of SGs on the mechanism of action of anti-cancer medications and was conducted using a six-stage methodological framework and the PRISMA guideline. Prior to October 2021, a systematic search of seven databases for eligible articles was performed. Following the review of the publications, the collected data were subjected to quantitative and qualitative analysis. Notably, Bortezomib, Sorafenib, Oxaliplatin, 5-fluorouracil, Cisplatin, and Doxorubicin accounted for the majority of the medications examined in the studies. Overall, this systematic scoping review attempts to demonstrate and give a complete overview of the function of SGs in the mechanism of action of anti-cancer medications by evaluating all research.
- Front Cell Dev Biol
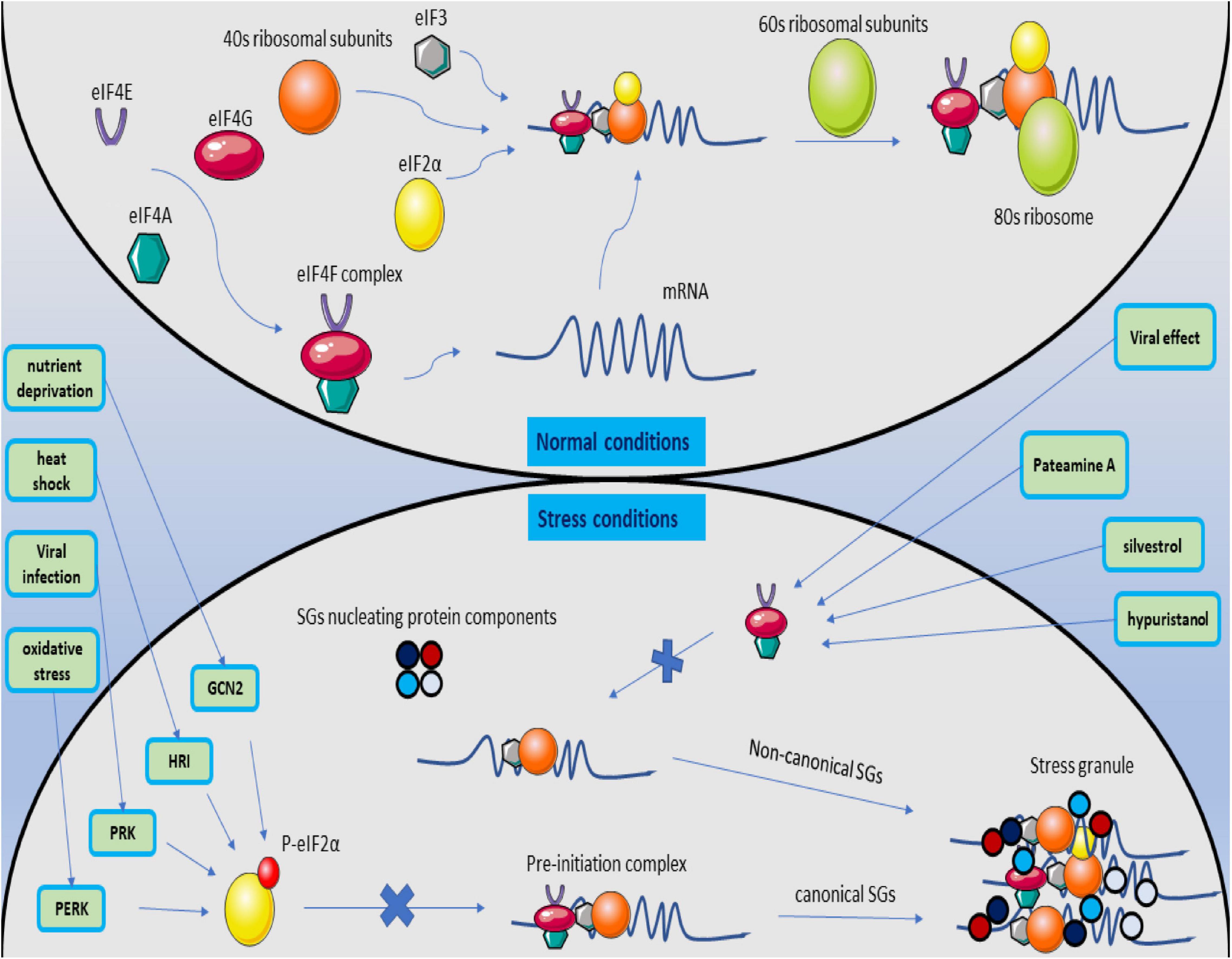 Stress Granules Involved in Formation, Progression and Metastasis of Cancer: A Scoping ReviewMohammad Reza Asadi, Dara Rahmanpour, Marziyeh Sadat Moslehian, and 5 more authors2021
Stress Granules Involved in Formation, Progression and Metastasis of Cancer: A Scoping ReviewMohammad Reza Asadi, Dara Rahmanpour, Marziyeh Sadat Moslehian, and 5 more authors2021The assembly of stress granules (SGs) is a well-known cellular strategy for reducing stress-related damage and promoting cell survival. SGs have become important players in human health, in addition to their fundamental role in the stress response. The critical role of SGs in cancer cells in formation, progression, and metastasis makes sense. Recent researchers have found that several SG components play a role in tumorigenesis and cancer metastasis via tumor-associated signaling pathways and other mechanisms. Gene-ontology analysis revealed the role of these protein components in the structure of SGs. Involvement in the translation process, regulation of mRNA stability, and action in both the cytoplasm and nucleus are among the main features of SG proteins. The present scoping review aimed to consider all studies on the effect of SGs on cancer formation, proliferation, and metastasis and performed based on a six-stage methodology structure and the PRISMA guideline. A systematic search of seven databases for qualified articles was conducted before July 2021. Publications were screened, and quantitative and qualitative analysis was performed on the extracted data. Go analysis was performed on seventy-one SGs protein components. Remarkably G3BP1, TIA1, TIAR, and YB1 have the largest share among the proteins considered in the studies. Altogether, this scoping review tries to demonstrate and provide a comprehensive summary of the role of SGs in the formation, progression, and metastasis of cancer by reviewing all studies.
- J Mol Neurosci
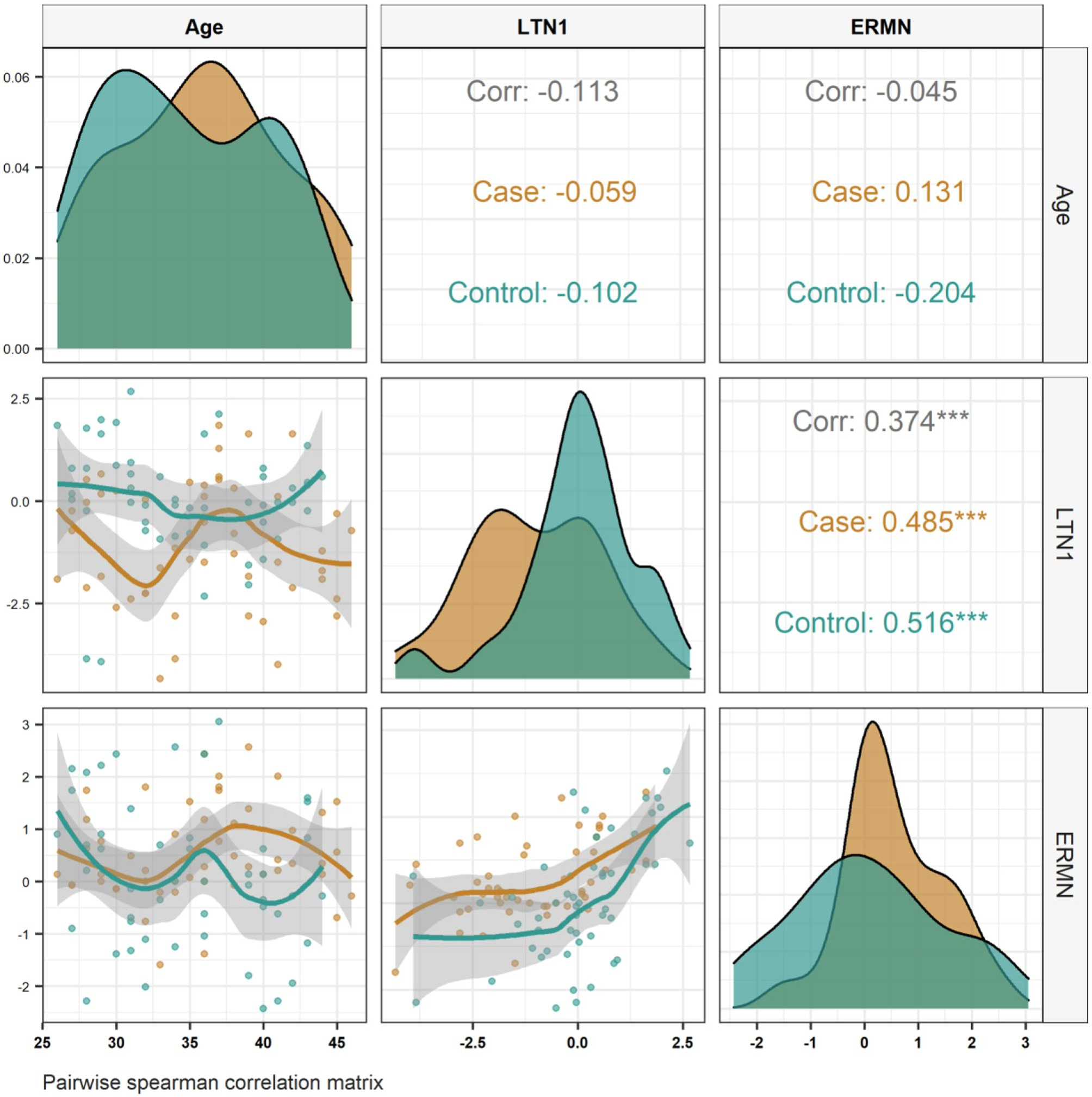 Expression Analysis of Ermin and Listerin E3 Ubiquitin Protein Ligase 1 Genes in the Periphery of Patients with SchizophreniaSara Farhang, Hani Sabaie, Jalal Gharesouran, and 5 more authors2021
Expression Analysis of Ermin and Listerin E3 Ubiquitin Protein Ligase 1 Genes in the Periphery of Patients with SchizophreniaSara Farhang, Hani Sabaie, Jalal Gharesouran, and 5 more authors2021Schizophrenia (SCZ) is a severe mental disorder with an unknown etiology. Recent researches indicate that correct myelination and translational regulation play a role in the pathogeny of SCZ. This study evaluated the expression pattern of Ermin (ERMN) and Listerin E3 ubiquitin protein ligase 1 (LTN1) genes, which play a role in myelination and ribosome quality control, respectively. The expression of the ERMN and LTN1 genes in the peripheral blood (PB) of 50 SCZ patients (male/female: 22/28, age (mean ± standard deviation (SD)): 35.9 ± 5.6) and 50 matched healthy controls (male/female: 23/27, age (mean ± SD): 34.7 ± 5.4) were assessed using quantitative polymerase chain reaction. Additionally, we used a bioinformatics approach based on microarray dataset analysis to examine the expression of these two genes in olfactory epithelium (OE) specimens. The expression of ERMN demonstrated no significant differences in PB samples among SCZ patients and healthy controls (adjusted P-value = 0.101). The expression of LTN1 was significantly higher in PB samples obtained from female patients compared with sex-matched controls (posterior beta = 1.734, adjusted P-value < 0.0001). Significant correlations were found between expression of the mentioned genes in PB samples both among SCZ patients and among healthy controls (r = 0.485, P < 0.001 and r = 0.516, P < 0.001, respectively). According to our in silico findings, the ERMN expression levels in OE samples of SCZ were statistically higher than those in controls (log2FC = 1.93, adj.P.Val = 9.66E-15). On the contrary, LTN1 expression levels in OE samples were statistically lower in SCZ cases versus controls (log2FC = − 0.77, adj.P.Val = 2.14E-06). Besides, a significant correlation was found between the expression of the mentioned genes in OE samples (r = − 0.60, P < 0.001). In conclusion, the present study is the first evidence to highlight the expression of the ERMN and LTN1 genes in the periphery of SCZ patients. Our findings may provide light on the SCZ’s pathogeny.
- Front Immunol
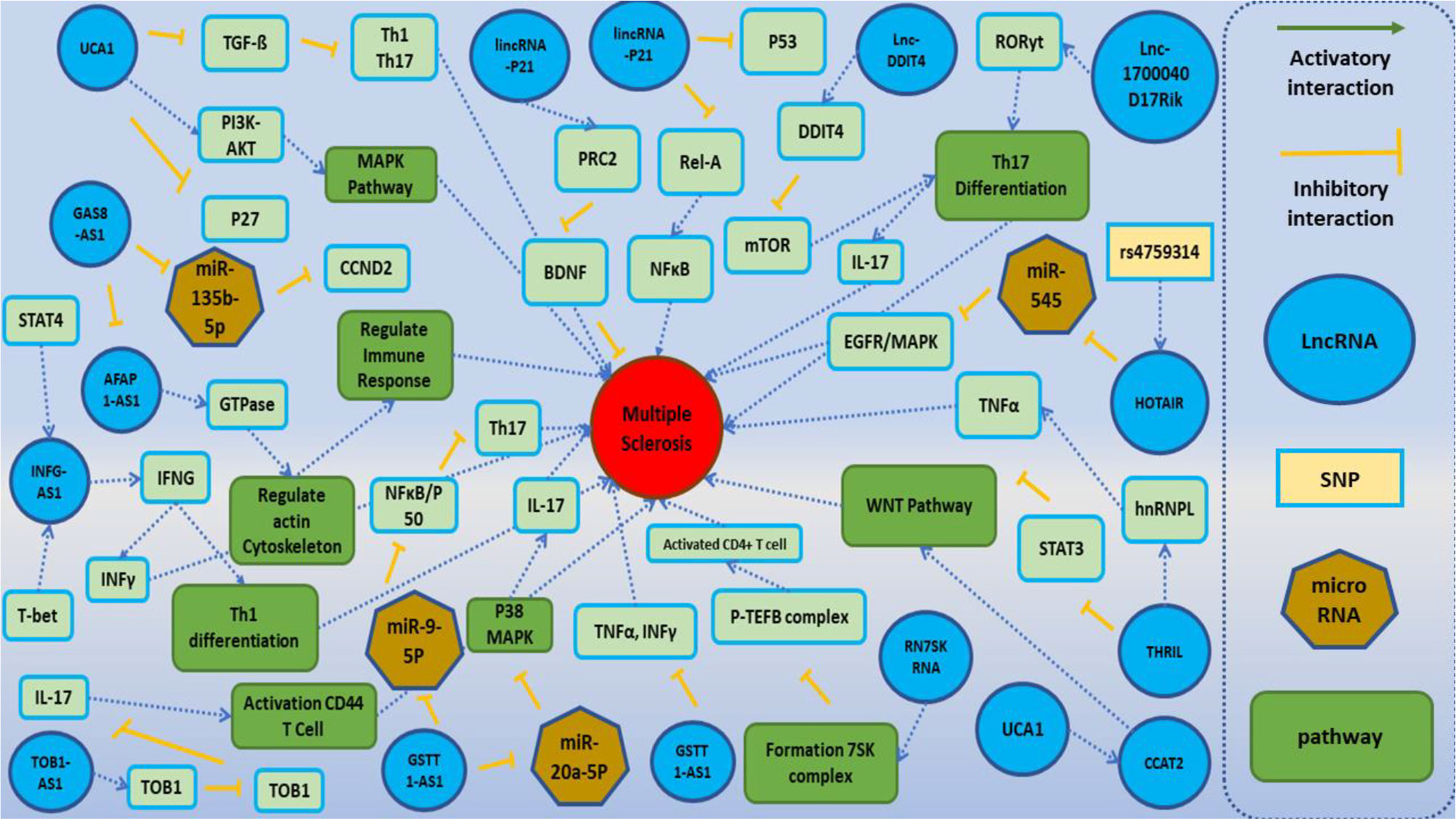 Long Non-Coding RNAs, Novel Offenders or Guardians in Multiple Sclerosis: A Scoping ReviewAbbas Jalaiei, Mohammad Reza Asadi, Hani Sabaie, and 6 more authors2021
Long Non-Coding RNAs, Novel Offenders or Guardians in Multiple Sclerosis: A Scoping ReviewAbbas Jalaiei, Mohammad Reza Asadi, Hani Sabaie, and 6 more authors2021Multiple sclerosis (MS), a chronic inflammatory demyelinating disease of the central nervous system, is one of the most common neurodegenerative diseases worldwide. MS results in serious neurological dysfunctions and disability. Disturbances in coding and non-coding genes are key components leading to neurodegeneration along with environmental factors. Long non-coding RNAs (lncRNAs) are long molecules in cells that take part in the regulation of gene expression. Several studies have confirmed the role of lncRNAs in neurodegenerative diseases such as MS. In the current study, we performed a systematic analysis of the role of lncRNAs in this disorder. In total, 53 studies were recognized as eligible for this systematic review. Of the listed lncRNAs, 52 lncRNAs were upregulated, 37 lncRNAs were downregulated, and 11 lncRNAs had no significant expression difference in MS patients compared with controls. We also summarized some of the mechanisms of lncRNA functions in MS. The emerging role of lncRNAs in neurodegenerative diseases suggests that their dysregulation could trigger neuronal death via still unexplored RNA-based regulatory mechanisms. Evaluation of their diagnostic significance and therapeutic potential could help in the design of novel treatments for MS.
- Front Aging Neurosci
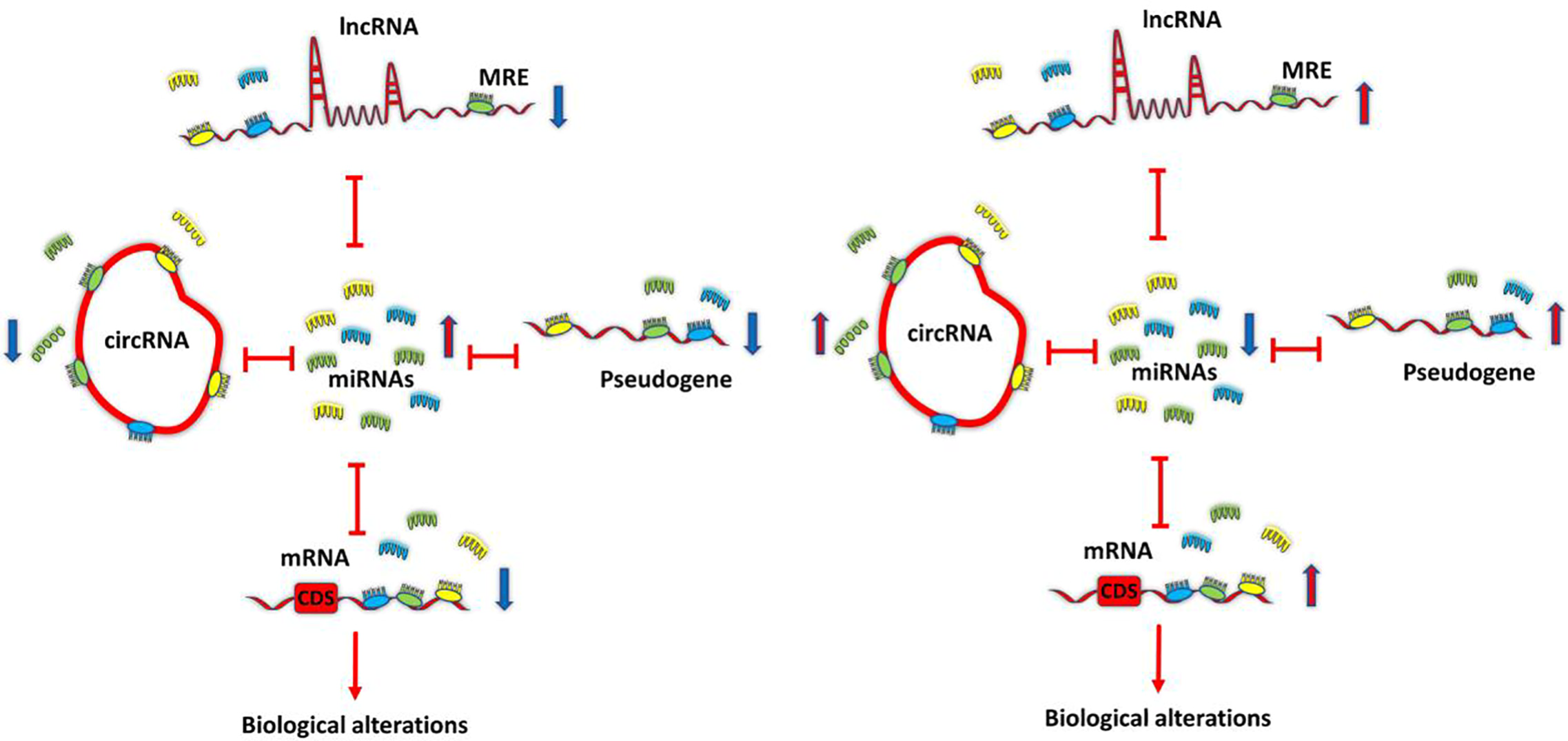 Molecular Insight Into the Therapeutic Potential of Long Non-coding RNA-Associated Competing Endogenous RNA Axes in Alzheimer’s Disease: A Systematic Scoping ReviewHani Sabaie, Nazanin Amirinejad, Mohammad Reza Asadi, and 5 more authors2021
Molecular Insight Into the Therapeutic Potential of Long Non-coding RNA-Associated Competing Endogenous RNA Axes in Alzheimer’s Disease: A Systematic Scoping ReviewHani Sabaie, Nazanin Amirinejad, Mohammad Reza Asadi, and 5 more authors2021Alzheimer’s disease (AD) is a heterogeneous degenerative brain disorder with a rising prevalence worldwide. The two hallmarks that characterize the AD pathophysiology are amyloid plaques, generated via aggregated amyloid β, and neurofibrillary tangle, generated via accumulated phosphorylated tau. At the post-transcriptional and transcriptional levels, the regulatory functions of non-coding RNAs, in particular long non-coding RNAs (lncRNAs), have been ascertained in gene expressions. It is noteworthy that a number of lncRNAs feature a prevalent role in their potential of regulating gene expression through modulation of microRNAs via a process called the mechanism of competing endogenous RNA (ceRNA). Given the multifactorial nature of ceRNA interaction networks, they might be advantageous in complex disorders (e.g., AD) investigations at the therapeutic targets level. We carried out scoping review in this research to analyze validated loops of ceRNA in AD and focus on ceRNA axes associated with lncRNA. This scoping review was performed according to a six-stage methodology structure and PRISMA guideline. A systematic search of seven databases was conducted to find eligible articles prior to July 2021. Two reviewers independently performed publications screening and data extraction, and quantitative and qualitative analyses were conducted. Fourteen articles were identified that fulfill the inclusion criteria. Studies with different designs reported nine lncRNAs that were experimentally validated to act as ceRNA in AD in human-related studies, including BACE1-AS, SNHG1, RPPH1, NEAT1, LINC00094, SOX21-AS1, LINC00507, MAGI2-AS3, and LINC01311. The BACE1-AS/BACE1 was the most frequent ceRNA pair. Among miRNAs, miR-107 played a key role by regulating three different loops. Understanding the various aspects of this regulatory mechanism can help elucidate the unknown etiology of AD and provide new molecular targets for use in therapeutic and clinical applications.
- Front Mol Biosci
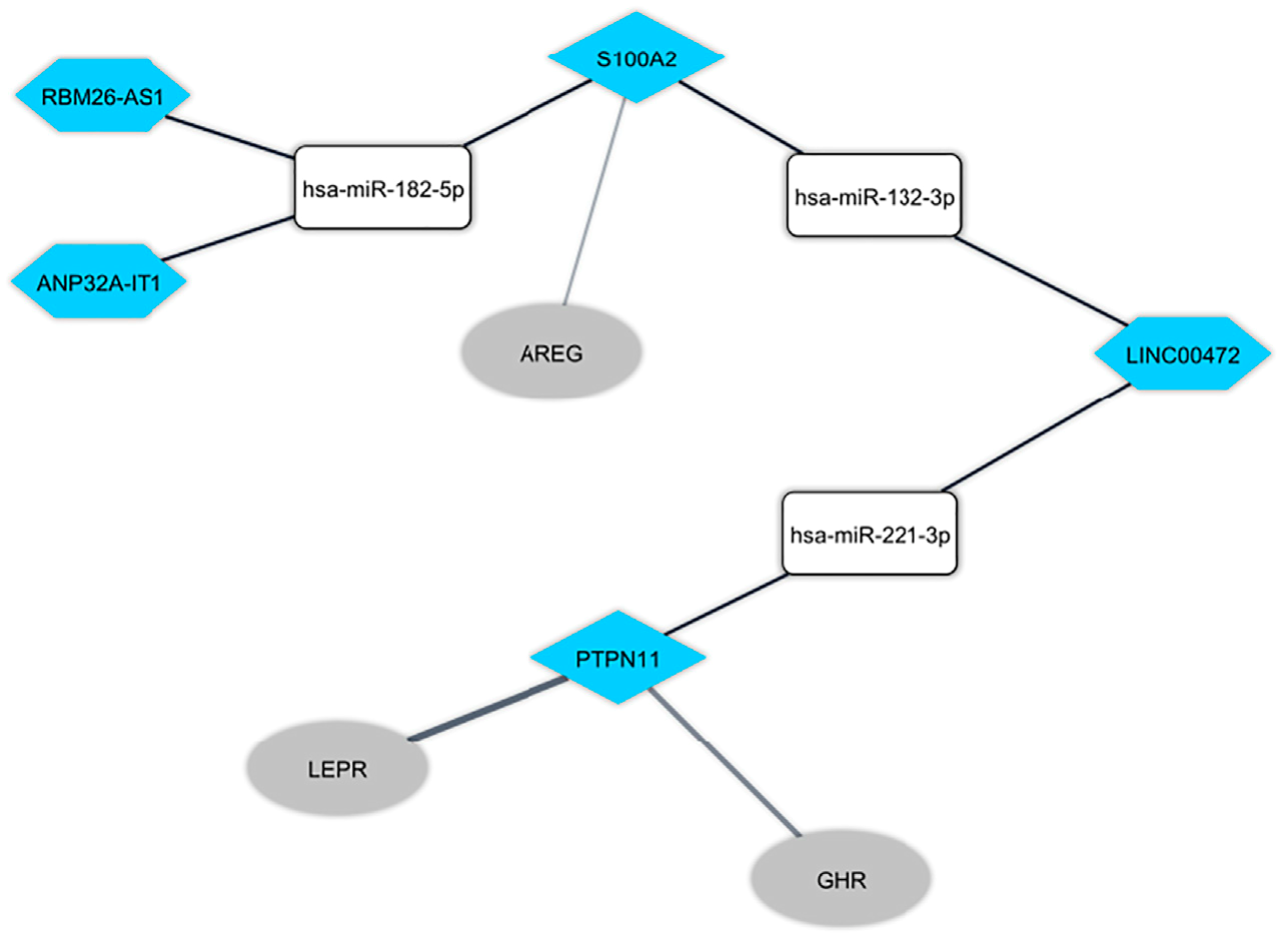 Mechanistic Insight Into the Regulation of Immune-Related Genes Expression in Autism Spectrum DisorderHani Sabaie, Hossein Dehghani, Shadi Shiva, and 4 more authors2021
Mechanistic Insight Into the Regulation of Immune-Related Genes Expression in Autism Spectrum DisorderHani Sabaie, Hossein Dehghani, Shadi Shiva, and 4 more authors2021Autism spectrum disorder (ASD) is a severe neurodevelopmental disorder featuring impairment in verbal and non-verbal interactions, defects in social interactions, stereotypic behaviors as well as restricted interests. In recent times, the incidence of ASD is growing at a rapid pace. In spite of great endeavors devoted to explaining ASD pathophysiology, its precise etiology remains unresolved. ASD pathogenesis is related to different phenomena associated with the immune system; however, the mechanisms behind these immune phenomena as well as the potential contributing genes remain unclear. In the current work, we used a bioinformatics approach to describe the role of long non-coding RNA (lncRNA)-associated competing endogenous RNAs (ceRNAs) in the peripheral blood (PB) samples to figure out the molecular regulatory procedures involved in ASD better. The Gene Expression Omnibus database was used to obtain the PB microarray dataset (GSE89594) from the subjects suffering from ASD and control subjects, containing the data related to both mRNAs and lncRNAs. The list of immune-related genes was obtained from the ImmPort database. In order to determine the immune-related differentially expressed mRNAs (DEmRNAs) and lncRNAs (DElncRNAs), the limma package of R software was used. A protein-protein interaction network was developed for the immune-related DEmRNAs. By employing the Human MicroRNA Disease Database, DIANA-LncBase, and DIANA-TarBase databases, the RNA interaction pairs were determined. We used the Pearson correlation coefficient to discover the positive correlations between DElncRNAs and DEmRNAs within the ceRNA network. Finally, the lncRNA-associated ceRNA network was created based on DElncRNA-miRNA-DEmRNA interactions and co-expression interactions. In addition, the KEGG enrichment analysis was conducted for immune-related DEmRNAs found within the constructed network. This work found four potential DElncRNA-miRNA-DEmRNA axes in ASD pathogenesis, including, LINC00472/hsa-miR-221-3p/PTPN11, ANP32A-IT1/hsa-miR-182-5p/S100A2, LINC00472/hsa-miR-132-3p/S100A2, and RBM26-AS1/hsa-miR-182-5p/S100A2. According to pathway enrichment analysis, the immune-related DEmRNAs were enriched in the “JAK-STAT signaling pathway” and “Adipocytokine signaling pathway.” An understanding of regulatory mechanisms of ASD-related immune genes would provide novel insights into the molecular mechanisms behind ASD pathogenesis.
- Sci Rep
 Long non-coding RNA-associated competing endogenous RNA axes in the olfactory epithelium in schizophrenia: a bioinformatics analysisHani Sabaie, Marziyeh Mazaheri Moghaddam, Madiheh Mazaheri Moghaddam, and 6 more authors2021
Long non-coding RNA-associated competing endogenous RNA axes in the olfactory epithelium in schizophrenia: a bioinformatics analysisHani Sabaie, Marziyeh Mazaheri Moghaddam, Madiheh Mazaheri Moghaddam, and 6 more authors2021The etiology of schizophrenia (SCZ), as a serious mental illness, is unknown. The significance of genetics in SCZ pathophysiology is yet unknown, and newly identified mechanisms involved in the regulation of gene transcription may be helpful in determining how these changes affect SCZ development and progression. In the current work, we used a bioinformatics approach to describe the role of long non-coding RNA (lncRNA)-associated competing endogenous RNAs (ceRNAs) in the olfactory epithelium (OE) samples in order to better understand the molecular regulatory processes implicated in SCZ disorders in living individuals. The Gene Expression Omnibus database was used to obtain the OE microarray dataset (GSE73129) from SCZ sufferers and control subjects, which contained information about both lncRNAs and mRNAs. The limma package of R software was used to identify the differentially expressed lncRNAs (DElncRNAs) and mRNAs (DEmRNAs). RNA interaction pairs were discovered using the Human MicroRNA Disease Database, DIANA-LncBase, and miRTarBase databases. In this study, the Pearson correlation coefficient was utilized to find positive correlations between DEmRNAs and DElncRNAs in the ceRNA network. Eventually, lncRNA-associated ceRNA axes were developed based on co-expression relations and DElncRNA-miRNA-DEmRNA interactions. This work found six potential DElncRNA-miRNA-DEmRNA loops in SCZ pathogenesis, including, SNTG2-AS1/hsa-miR-7-5p/SLC7A5, FLG-AS1/hsa-miR-34a-5p/FOSL1, LINC00960/hsa-miR-34a-5p/FOSL1, AQP4-AS1/hsa-miR-335-5p/FMN2, SOX2-OT/hsa-miR-24-3p/NOS3, and CASC2/hsa-miR-24-3p/NOS3. According to the findings, ceRNAs in OE might be promising research targets for studying SCZ molecular mechanisms. This could be a great opportunity to examine different aspects of neurodevelopment that may have been hampered early in SCZ patients.
- Front Immunol
 Long Non-Coding RNA- Associated Competing Endogenous RNA Axes in T-Cells in Multiple SclerosisHani Sabaie, Zoha Salkhordeh, Mohammad Reza Asadi, and 6 more authors2021
Long Non-Coding RNA- Associated Competing Endogenous RNA Axes in T-Cells in Multiple SclerosisHani Sabaie, Zoha Salkhordeh, Mohammad Reza Asadi, and 6 more authors2021Multiple sclerosis (MS) is an immune-mediated demyelinating and degenerative disease with unknown etiology. Inappropriate response of T-cells to myelin antigens has an essential role in the pathophysiology of MS. The clinical and pathophysiological complications of MS necessitate identification of potential molecular targets to understand the pathogenic events of MS. Since the functions and regulatory mechanisms of long non-coding RNAs (lncRNAs) acting as competing endogenous RNAs (ceRNAs) in MS are yet uncertain, we conducted a bioinformatics analysis to explain the lncRNA-associated ceRNA axes to clarify molecular regulatory mechanisms involved in T-cells responses in MS. Two microarray datasets of peripheral blood T-cell from subjects with relapsing-remitting MS and matched controls containing data about miRNAs (GSE43590), mRNAs and lncRNAs (GSE43591) were downloaded from the Gene Expression Omnibus database. Differentially expressed miRNAs (DEmiRNAs), mRNAs (DEmRNAs), and lncRNAs (DElncRNAs) were identified by the limma package of the R software. Protein-protein interaction (PPI) network and module were developed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) and the Molecular Complex Detection (MCODE) Cytoscape plugin, respectively. Using DIANA-LncBase and miRTarBase, the lncRNA-associated ceRNA axes was constructed. We conducted a Pearson correlation analysis and selected the positive correlations among the lncRNAs and mRNAs in the ceRNA axes. Lastly, DEmRNAs pathway enrichment was conducted by the Enrichr tool. A ceRNA regulatory relationship among Small nucleolar RNA host gene 1 (SNHG1), hsa-miR-197-3p, YOD1 deubiquitinase (YOD1) and zinc finger protein 101 (ZNF101) and downstream connected genes was identified. Pathway enrichment analysis showed that DEmRNAs were enriched in “Protein processing in endoplasmic reticulum” and “Herpes simplex virus 1 infection” pathways. To our knowledge, this would be the first report of a possible role of SNHG1/hsa-miR-197-3p/YOD1/ZNF101 axes in the pathogenesis of MS. This research remarks on the significance of ceRNAs and prepares new perceptions for discovering the molecular mechanism of MS.
- Sci Rep
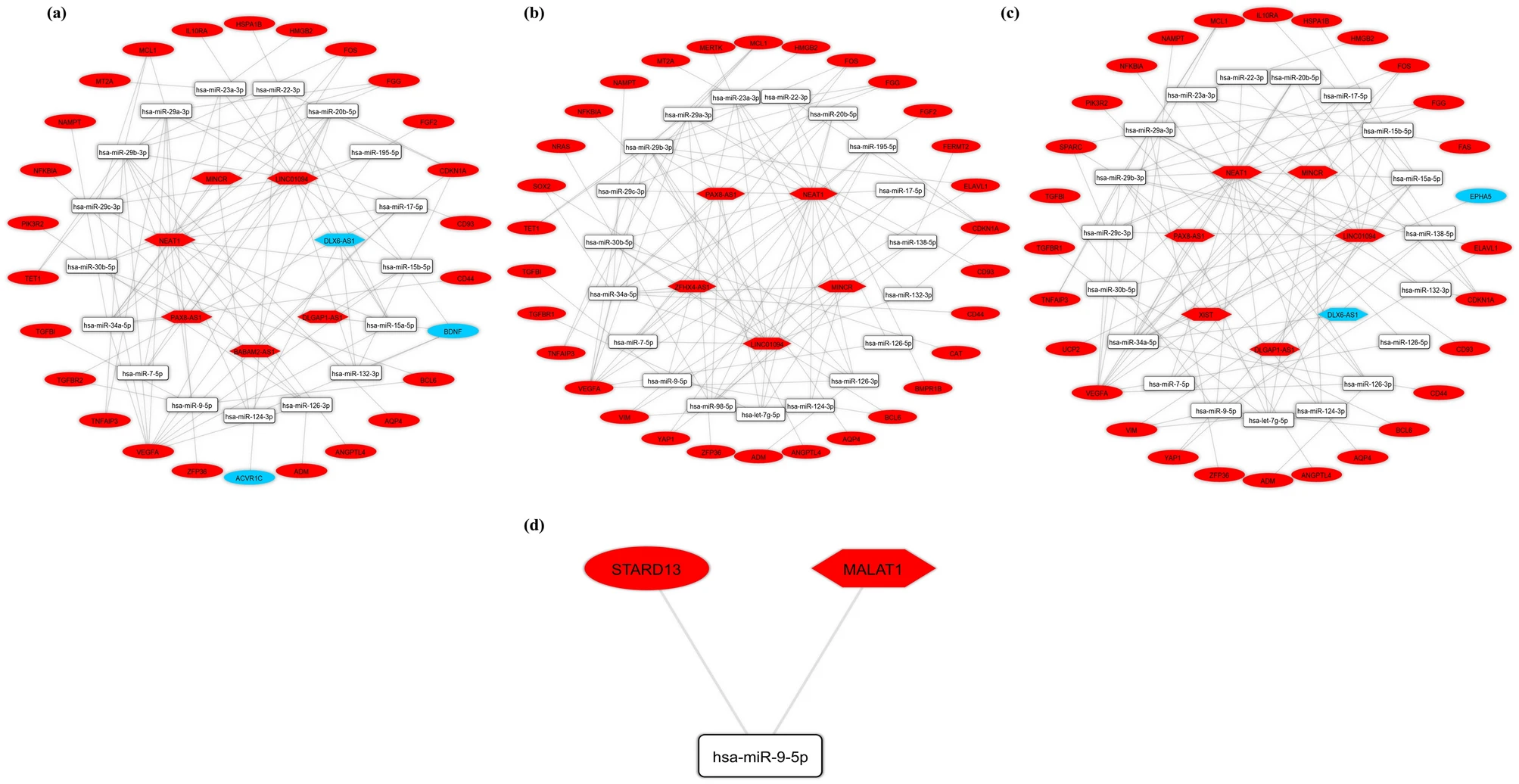 Bioinformatics analysis of long non-coding RNA-associated competing endogenous RNA network in schizophreniaHani Sabaie, Madiheh Mazaheri Moghaddam, Marziyeh Mazaheri Moghaddam, and 5 more authors2021
Bioinformatics analysis of long non-coding RNA-associated competing endogenous RNA network in schizophreniaHani Sabaie, Madiheh Mazaheri Moghaddam, Marziyeh Mazaheri Moghaddam, and 5 more authors2021Schizophrenia (SCZ) is a serious psychiatric condition with a 1% lifetime risk. SCZ is one of the top ten global causes of disabilities. Despite numerous attempts to understand the function of genetic factors in SCZ development, genetic components in SCZ pathophysiology remain unknown. The competing endogenous RNA (ceRNA) network has been demonstrated to be involved in the development of many kinds of diseases. The ceRNA hypothesis states that cross-talks between coding and non-coding RNAs, including long non-coding RNAs (lncRNAs), via miRNA complementary sequences known as miRNA response elements, creates a large regulatory network across the transcriptome. In the present study, we developed a lncRNA-related ceRNA network to elucidate molecular regulatory mechanisms involved in SCZ. Microarray datasets associated with brain regions (GSE53987) and lymphoblasts (LBs) derived from peripheral blood (sample set B from GSE73129) of SCZ patients and control subjects containing information about both mRNAs and lncRNAs were downloaded from the Gene Expression Omnibus database. The GSE53987 comprised 48 brain samples taken from SCZ patients (15 HPC: hippocampus, 15 BA46: Brodmann area 46, 18 STR: striatum) and 55 brain samples taken from control subjects (18 HPC, 19 BA46, 18 STR). The sample set B of GSE73129 comprised 30 LB samples (15 patients with SCZ and 15 controls). Differentially expressed mRNAs (DEmRNAs) and lncRNAs (DElncRNAs) were identified using the limma package of the R software. Using DIANA-LncBase, Human MicroRNA Disease Database (HMDD), and miRTarBase, the lncRNA- associated ceRNA network was generated. Pathway enrichment of DEmRNAs was performed using the Enrichr tool. We developed a protein–protein interaction network of DEmRNAs and identified the top five hub genes by the use of STRING and Cytoscape, respectively. Eventually, the hub genes, DElncRNAs, and predictive miRNAs were chosen to reconstruct the subceRNA networks. Our bioinformatics analysis showed that twelve key DEmRNAs, including BDNF, VEGFA, FGF2, FOS, CD44, SOX2, NRAS, SPARC, ZFP36, FGG, ELAVL1, and STARD13, participate in the ceRNA network in SCZ. We also identified DLX6-AS1, NEAT1, MINCR, LINC01094, DLGAP1-AS1, BABAM2-AS1, PAX8-AS1, ZFHX4-AS1, XIST, and MALAT1 as key DElncRNAs regulating the genes mentioned above. Furthermore, expression of 15 DEmRNAs (e.g., ADM and HLA-DRB1) and one DElncRNA (XIST) were changed in both the brain and LB, suggesting that they could be regarded as candidates for future biomarker studies. The study indicated that ceRNAs could be research candidates for investigating SCZ molecular pathways.
- Front Mol Neurosci
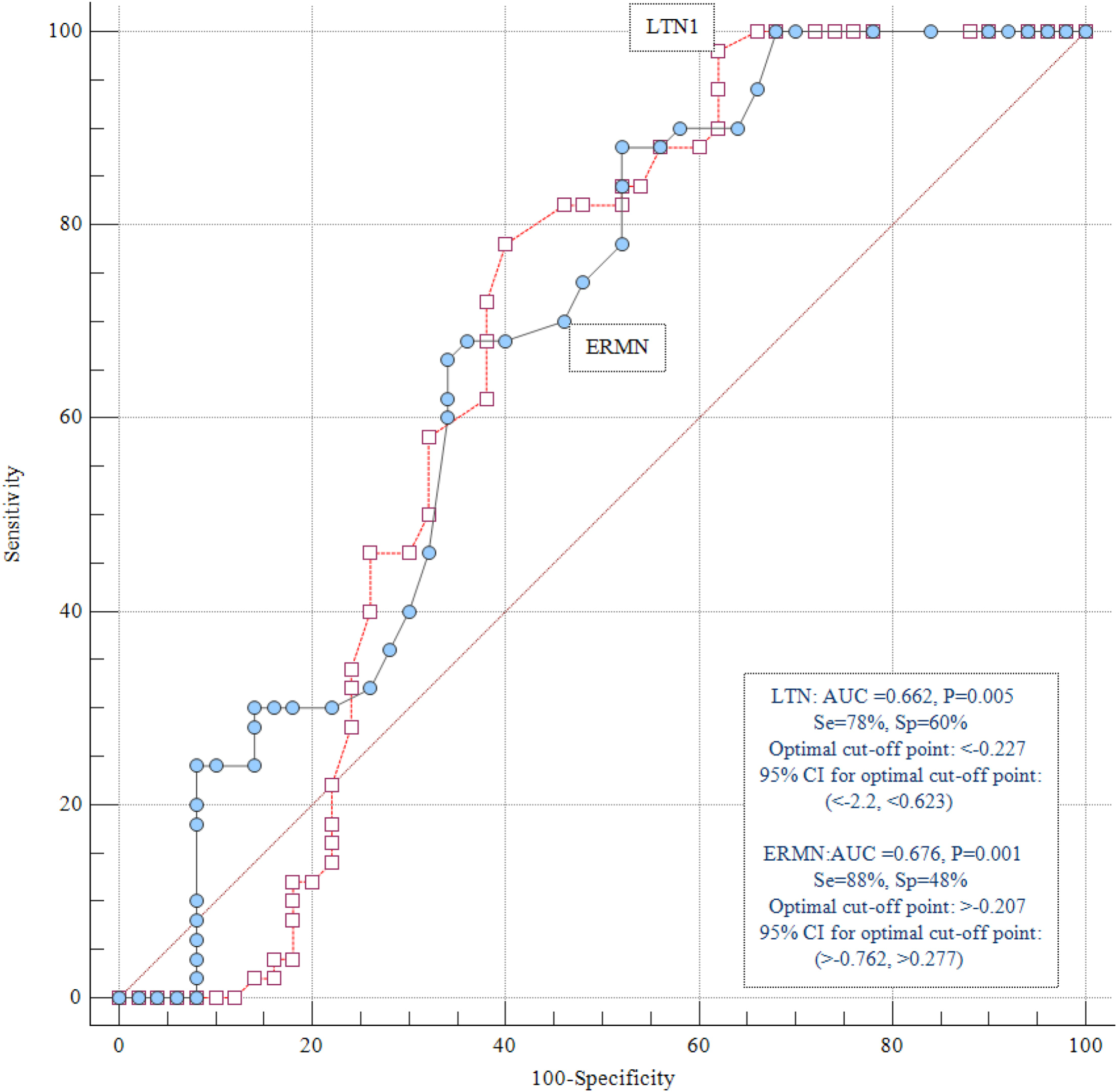 Expression Analysis of Ermin and Listerin E3 Ubiquitin Protein Ligase 1 Genes in Autistic PatientsShadi Shiva, Jalal Gharesouran, Hani Sabaie, and 4 more authors2021
Expression Analysis of Ermin and Listerin E3 Ubiquitin Protein Ligase 1 Genes in Autistic PatientsShadi Shiva, Jalal Gharesouran, Hani Sabaie, and 4 more authors2021Autism spectrum disorder (ASD) is a severe neurodevelopmental disorder that involves social interaction defects, impairment of non-verbal and verbal interactions, and limited interests along with stereotypic activities. Its incidence has been increasing rapidly in recent decades. Despite numerous attempts to understand the pathophysiology of ASD, its exact etiology is still unclear. Recent data shows the role of accurate myelination and translational regulation in ASD’s pathogenesis. In this study, we assessed Ermin (ERMN) and Listerin E3 Ubiquitin Protein Ligase 1 (LTN1) genes expression in Iranian ASD patients and age- and gender-matched healthy subjects’ peripheral blood using quantitative real-time PCR to recognize any probable dysregulation in the expression of these genes and propose this disorder’s mechanisms. Analysis of the expression demonstrated a significant ERMN downregulation in total ASD patients compared to the healthy individuals (posterior beta = −0.794, adjusted P-value = 0.025). LTN1 expression was suggestively higher in ASD patients in comparison with the corresponding control individuals. Considering the gender of study participants, the analysis showed that the mentioned genes’ different expression levels were significant only in male subjects. Besides, a significant correlation was found between expression of the mentioned genes (r = −0.49, P < 0.0001). The present study provides further supports for the contribution of ERMN and LTN1 in ASD’s pathogenesis.
2020
- Biomed Pharmacother
 Clinical and genetic features of PEHO and PEHO-Like syndromes: A scoping reviewHani Sabaie, Noora Karim Ahangar, Soudeh Ghafouri-Fard, and 2 more authors2020
Clinical and genetic features of PEHO and PEHO-Like syndromes: A scoping reviewHani Sabaie, Noora Karim Ahangar, Soudeh Ghafouri-Fard, and 2 more authors2020Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO) syndrome is a genetic neurological condition characterized by extreme cerebellar atrophy. PEHO-Like syndrome is comparable to PEHO syndrome, with the exception that there is no typical neuro-radiologic or neuro-ophthalmic findings. PEHO spectrum disorders are highly clinically and genetically heterogeneous, and this has challenged their diagnosis. This scoping review aims to summarize and discuss common clinical and genetic features of these syndromes to help future researches. This study was performed according to a six-stage methodology structure and PRISMA guideline. A systematic search of seven databases was performed to find eligible publications prior to June 2020. Articles screening and data extraction were independently performed by two reviewers and quantitative and qualitative analyses were conducted. Thirty-eight articles were identified that fulfill the inclusion criteria. Cerebellar atrophy was the main clinical difference between the two groups but data on optic atrophy and infantile spasms/hypsarrhythmia were not consistent with the previously essential diagnostic criteria. Genetic analysis was performed in several studies, leading to identification of pathogenic variants in different genes that caused these conditions due to different mechanisms. Genetic studies could revolutionize the diagnosis process and our understanding of the etiology of this challenging group of patients by providing targeted sequencing panels and exome- or genome-scale studies in the future.